论文阅读笔记
Abstract
squencing-based spatial transcriptomics(ST)技术无法实现单细胞分辨率,而通过单细胞RNA测序的数据与ST数据存在偏差,本文开发了一个基于实例的迁移学习框架,通过ST数据调整scRNA-seq数据,从而更精确地描述细胞类型空间组织
Introduction
SPOTlight:基于非负矩阵分解回归,缺点是无法区分个体细胞类型地贡献
SpatialDWLS:增强了特异性但消除了不想管细胞类型影响,并可能过滤稀有类型
CARD:可以在scRNAseq数据不匹配地情况下分解但没有奥利scRNA-seq和ST数据的差异
stereoscope假设scRNA和ST基于二项分布
RCTD利用泊松分布,假设细胞类型的共享随机效应。
Cell2location采用分层贝叶斯后验分布近似ST数据,拟合性差。
STRIDE:基于潜在狄利克雷分布
DSTG:基于图卷积网络。
大多数方法按时scRNA和ST数据分布相似,但很少考虑差异
本文设计的基于实例的迁移学习框架通过ST数据调整scRNA-seq数据,以改进分解,采用核平均匹配方法
Results
scRNA-seq和ST数据的分布差异
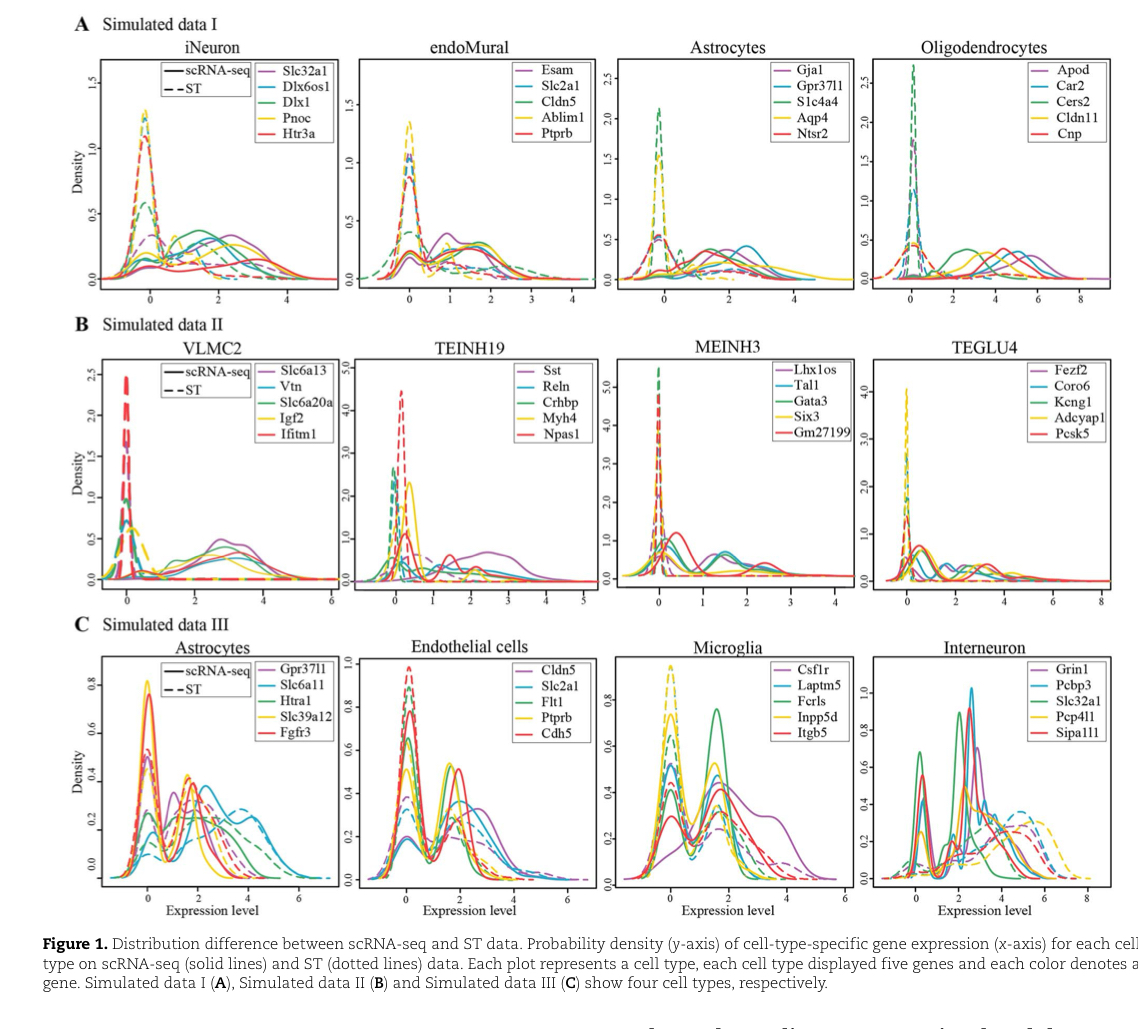
有三个模拟数据集,为了更好的说明不同技术导致的分布差异存在,
采用Simulated Data III .对于每种细胞类型,我们检查属于该类型的细胞中细胞类型特异性基因的表达水平,然后比较相同基因在scRNA-seq和ST数据中的概率密度分布。
对于三个模拟数据的每种细胞类型绘制出概率密度曲线,能够直观的看出模拟数据三的优越性
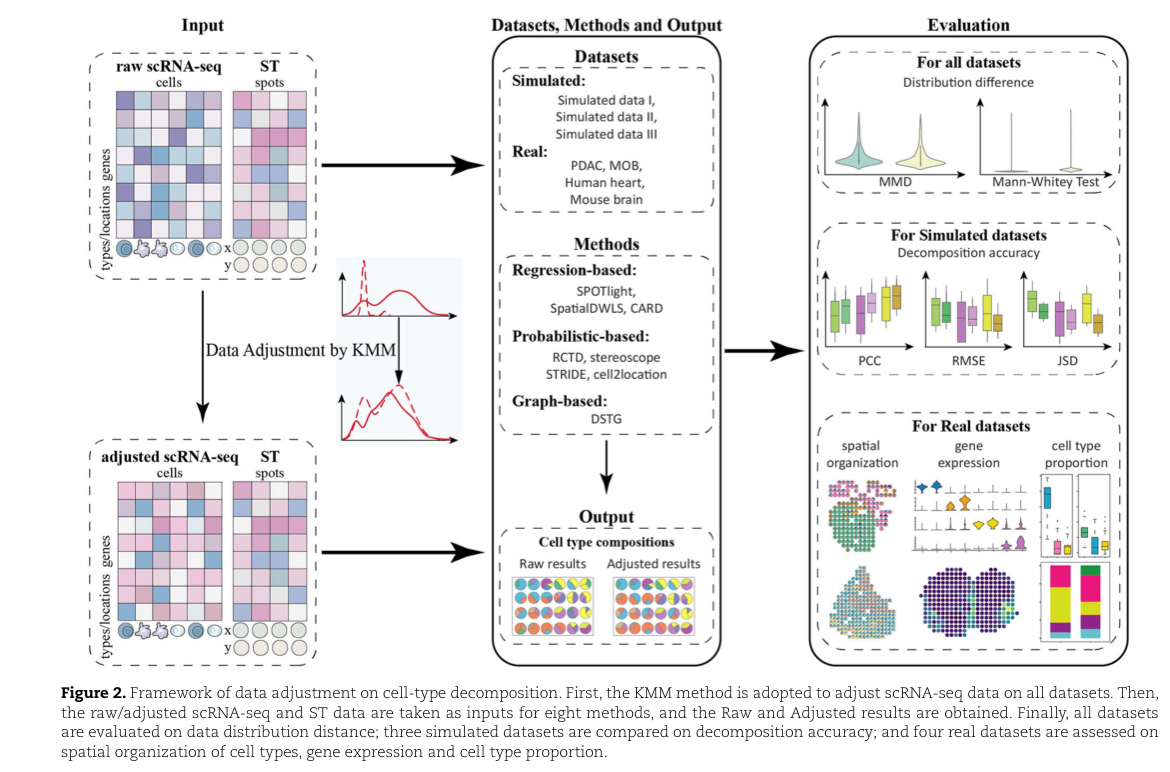
数据调整框架
首先采用KMM方法,通过ST调整scRNA数据,KMM改变了每种细胞类型中相应细胞在相应细胞类型特异性基因上的权重。
然后用三个模拟数据和四个真实数据套入模型,去关注它们的性质。
最后将Raw结果和调整后的结果比较
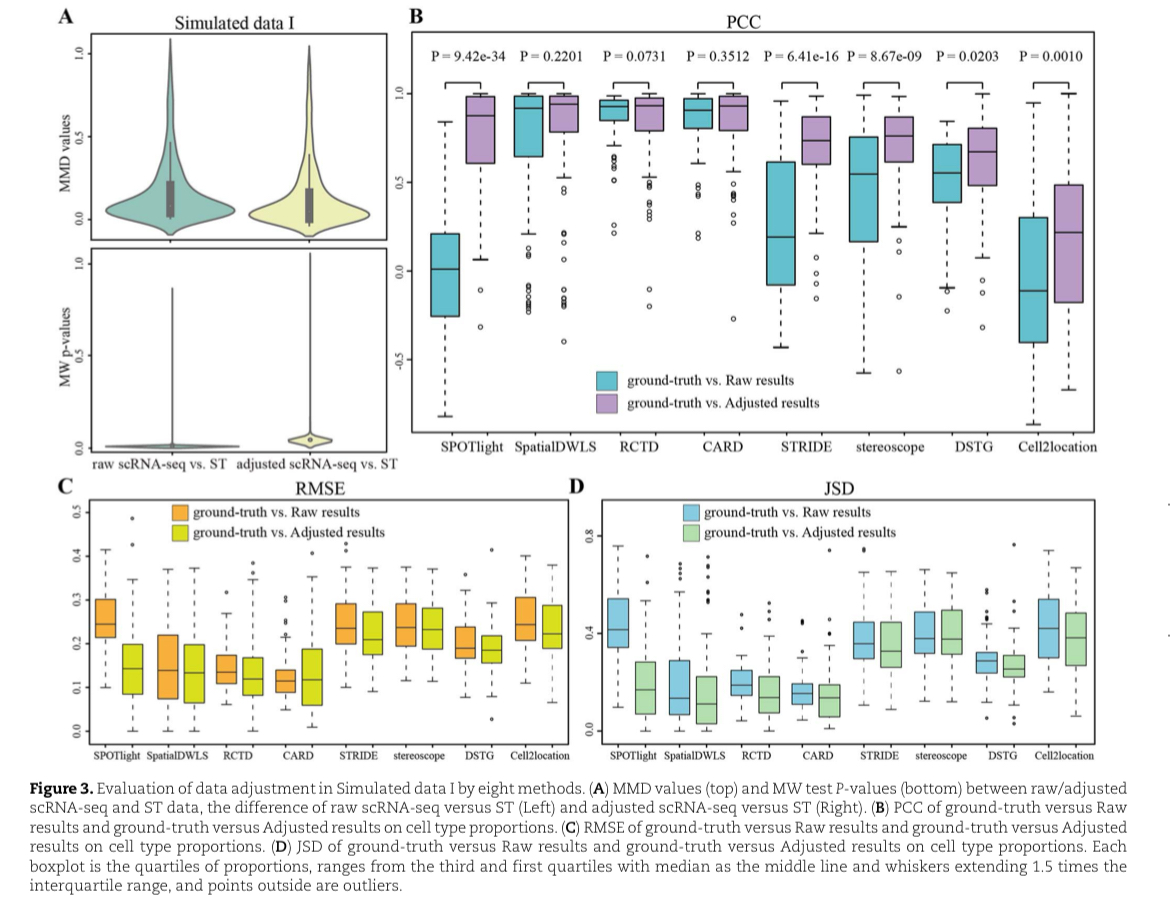
评估调整数据与模拟数据
三个模拟数据集提供了scRNA-seq数据调整提高ST数据分解精度的证据,分解结果容易收到两个数据之间分布差异的影响
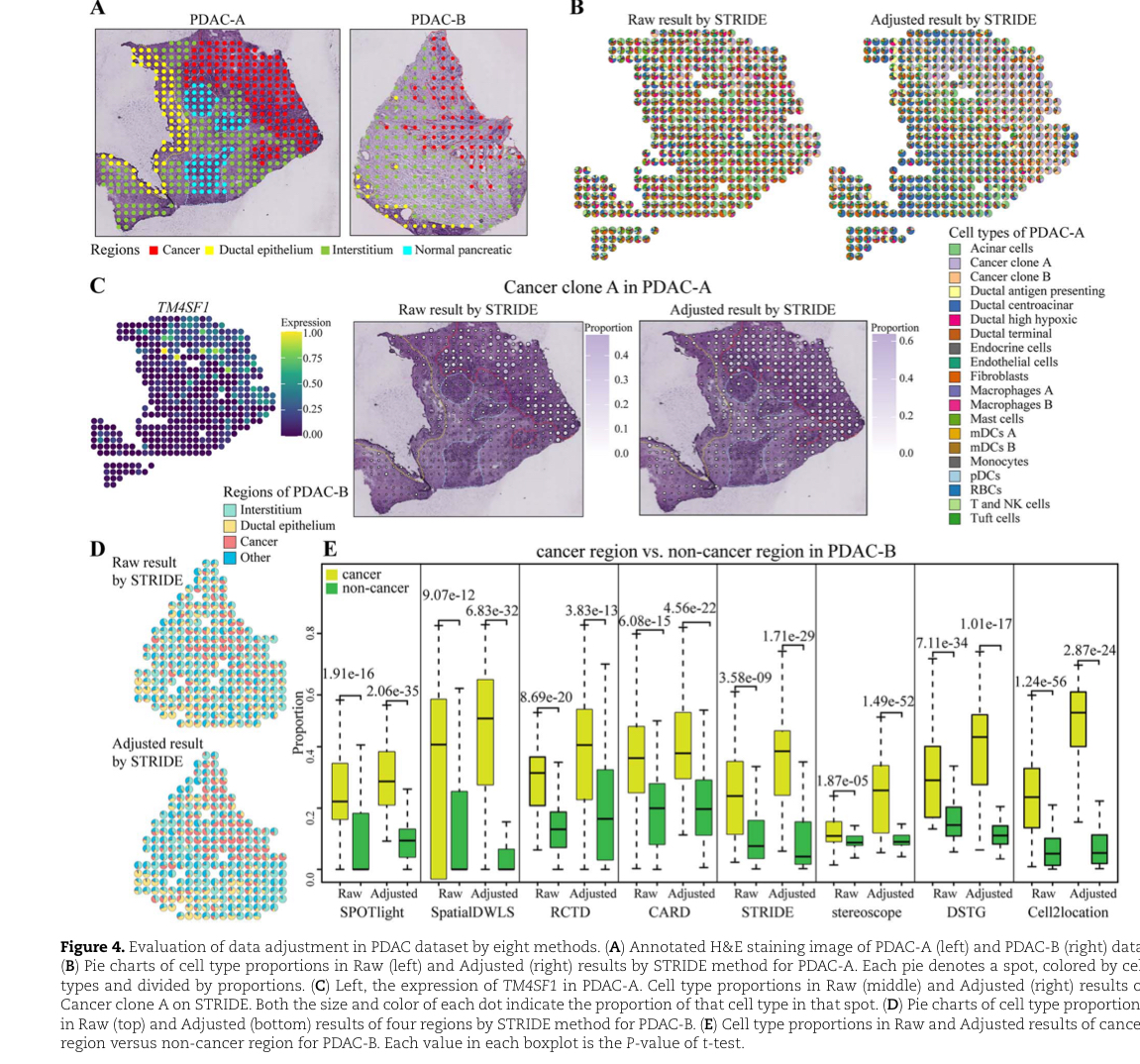
PDAC数据集上癌细胞类型的空间描述
PDAC数据表明,数据调整改善了癌细胞类型的空间描述,对细胞类型的空间组织进行更精细的注释,可以直接使用低分辨率ST数据来描绘癌症样本。
在人类心脏上的表现
数据调整使某些基因与ST数据的相关性增强,并改进了分解,更丰富的基因有助于我们研究心脏相关疾病
##在小鼠嗅球数据集上的表现
调整后的数据的表达模式显示出明显的细胞类型和层的空间组织。
Methods
剩下的就是理论推导,大致这篇论文想干什么,怎么干的已经了解了,后面就不写了。