Title
Virtual Cell Challenge:
Toward a Turing test for the virtual cell
Introduction
These
‘‘virtual cells’’ are expected to learn the
relationship between cell state and function
and are intended to predict the conse
quences of perturbations—such as a
gene knockdown or the application of a
drug—across cell types and cell contexts.
模型需要考虑到的因素:These models must account for
additional complexity—such as the cell
type, genetic background, and context of
a cell—as well as the cellular phenotype
being measured and predicted.
Open-source competitions can lead to rapid progress
遇到的问题:Without standard
ized benchmarks and purpose-built
evaluation datasets that evolve in real
time alongside developments in the field,
it is difficult to evaluate whether models
are capturing generalizable biological
structure rather than dataset-specific
patterns.
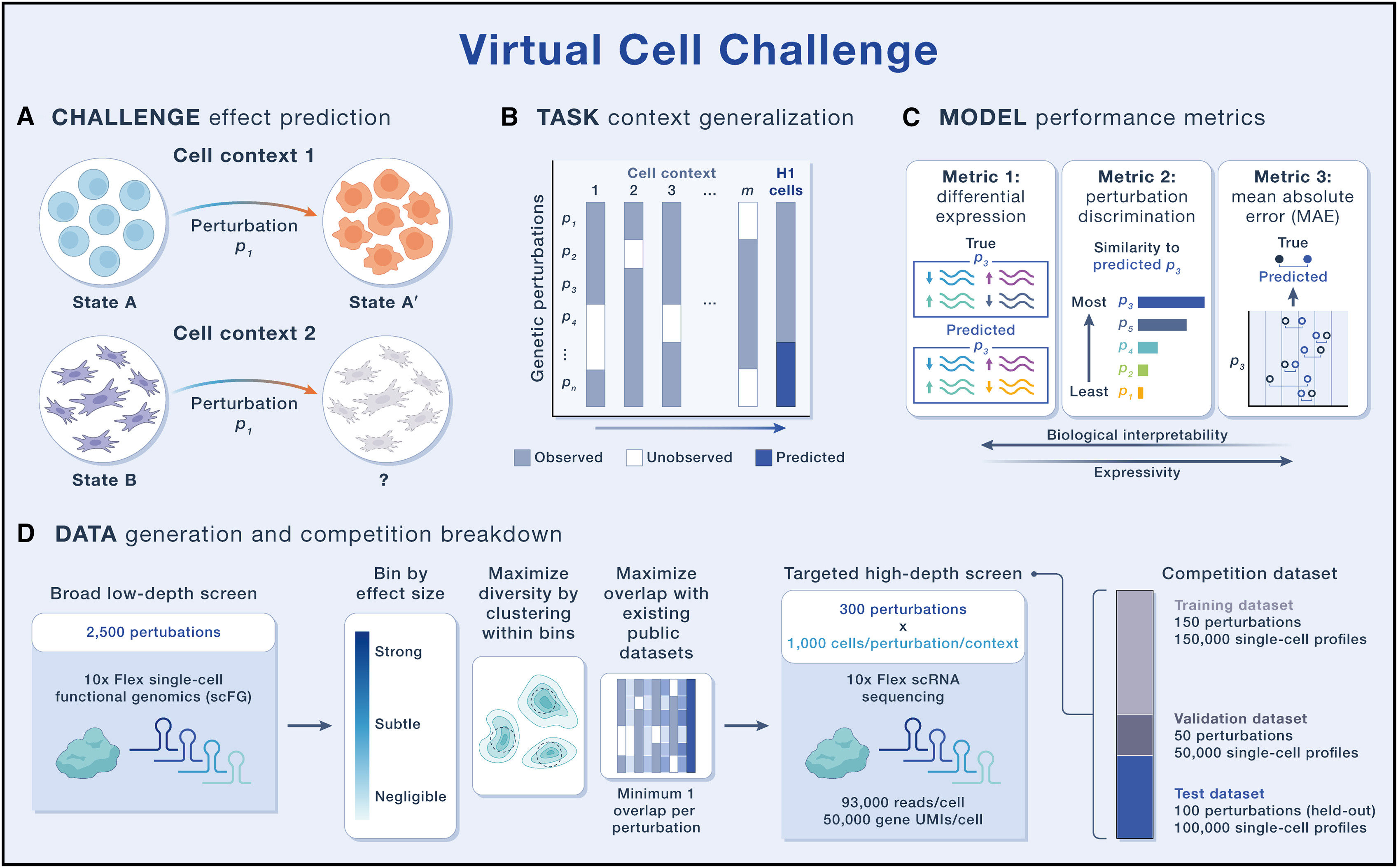
Datasets
human embryonic stem cell line (H1 hESC)
scFG to
generate approximately 300,000 single-cell RNA-sequencing (scRNA-seq) profiles by silencing 300 carefully selected genes using CRISPR interference (CRISPRi)
Format of the Virtual Cell Challenge Task
Predictive models can be trained to generalize along several axes.
- (1) generalization across biological context (e.g., cell type, cell line, culture conditions, or even in vivo versus in vitro settings)
- (2) generalization to novel genetic and/or chemical perturbations, including their combinations.
这里采用的形式是针对cell type进行预测,考虑到zero-shot的不切实际,采取few-shot作为训练模型的手段
Evaluations
Evaluation metrics should reflect the core purpose of a virtual cell: simulating cellular behavior via in silico experiments—specifically, predicting gene expression responses to genetic perturbations.
- The differential expression score evaluates how accurately a model predicts differential gene expression, a key output of most scFG exoeriments and an essential input for downstream biological interpretation.
- The perturbation discrimination score
measures a model’s ability to distinguish
between perturbations by ranking predic
tions according to their similarity to the
true perturbational effect, regardless of
their effect size.
模型可能投机取巧:
- 如果只会输出固定的基因集 → DE 得分高,但扰动区分能力差。
- 如果只会在 embedding 空间里分组 → 扰动区分能力强,但 DE 无生物学意义。
mean absolute
error (MAE). While MAE is less biologically
interpretable, it captures overall predic
tive accuracy and provides a global view
of model performance across the entire
gene expression profile.