scPerturb: harmonized single-cell perturbation data.
第一次接触这个领域,仔细的研读一下。
# Abstract
这篇文章应用了 uniform quality control pipelines 和 harmonize feature annotations. 然后对于它的模型 scPerturb 的目的是 facilitates comparison and integration across datasets. 它们描述了一个 energy statistics (E-statistics) 去衡量 perturbation 和显著性检验。
This work provides an information resource for researchers working with single-cell perturbation data and recommendations for experimental design, including optimal cell counts and read depth.
# Introduction
Perturbation experiments probe the response of cells or cellular systems to changes in conditions.
Nowadays, with advanced functional genomics techniques, single-cell genetic perturbations acting on individual cellular components are available. Perturbations using different technologies target different layers of the hierarchy of protein production.
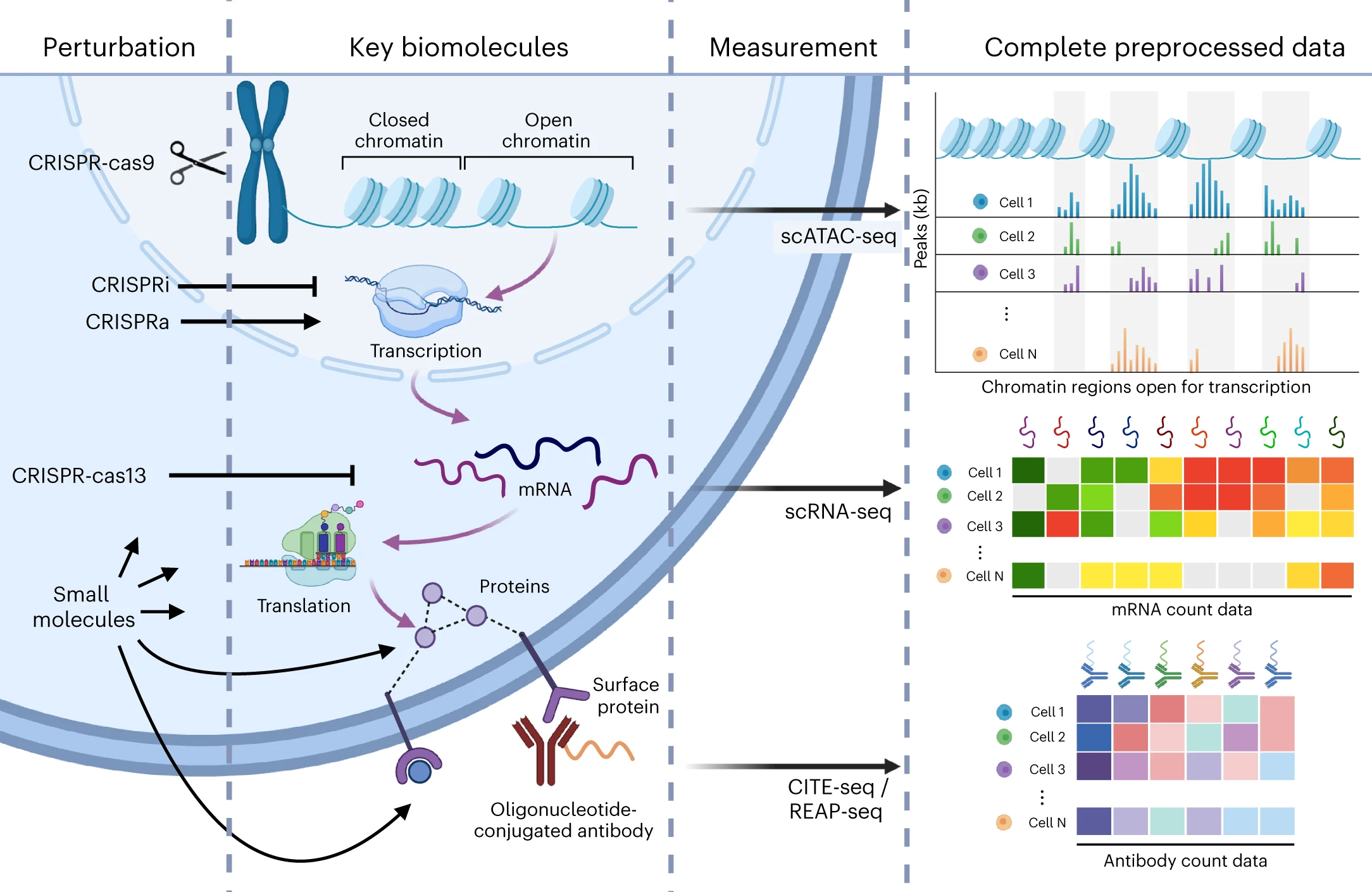
这里解释一下这张图,上面提到的 different technologies 指的是例如 CRISPR-cas9、CRISPRi 这些东西。比如 CRISPR-cas9 它在基因上直接做增删,CRISPRi 和 CRISPRa 分别抑制和激活转录 (transcription),CRISPR-cas13 切割 RNA,一些小分子的药物影响蛋白质产物例如酶和受体。
然后是比较关键的一句话: Barcodes associated with unique CRISPR guide perturbations are read alongside single-cell RNA sequencing (scRNA-seq), CITE-seq (cellular indexing of transcriptomes and epitopes by sequencing) or scATAC-seq (single-cell sequencing assay for transposase-accessible chromatin) to identify each cell’s perturbation condition.
感觉很难理解上面这句话,我拿 GPT 跑了一个解释。
三种数据
下面分别对这三种单细胞多组学技术做详细介绍,包括它们的基本原理、实验流程、优势与局限,以及典型应用场景。
# 1. Single-cell RNA sequencing (scRNA-seq)
# 基本原理
scRNA-seq 用于在单细胞水平上定量每个细胞的转录组(mRNA)表达。它能够揭示细胞间在基因表达谱上的异质性,识别不同细胞类型、细胞状态和发育轨迹。
# 实验流程
单细胞分离
- 微流控芯片(如 10x Genomics Chromium)或滴式微流控(Drop-seq、inDrop)将单个细胞包封于微小液滴或孔室中。
细胞裂解与逆转录
- 每个液滴内加入含有细胞条形码(cell barcode)和分子标签(UMI, unique molecular identifier)的引物,裂解细胞并逆转录生成带有条形码的 cDNA。
文库构建与测序
- 将所有液滴中的 cDNA 汇合,进行扩增、建库并上机高通量测序(Illumina)。
数据处理与分析
- 根据条形码将每条读数(read)分配回对应细胞,UMI 去重后得到每个细胞中每个基因的表达量矩阵。
- 下游分析包括细胞聚类、差异表达、伪时序分析等。
# 优势
- 能在单细胞水平揭示生物体系的异质性。
- 支持大规模细胞通量(可达数万到数十万细胞)。
- 数据成熟,分析工具丰富(Seurat、Scanpy 等)。
# 局限
- 只捕获多聚 A 尾 mRNA,无法直接测到非 polyadenylated RNA(如部分非编码 RNA)。
- 有 “高掉落率”(dropout)现象:低丰度转录本常常检测不到。
- 无空间信息,需要结合空间转录组或成像技术。
# 应用场景
- 细胞类型鉴定与注释
- 发育过程伪时序重构
- 疾病微环境中的免疫细胞分型
- 药物或基因扰动后细胞响应分析
# 2. CITE-seq
(Cellular Indexing of Transcriptomes and Epitopes by Sequencing)
# 基本原理
CITE-seq 在常规模版 scRNA-seq 的基础上,额外对细胞表面或胞内蛋白进行标记与测量。它使用带有寡核苷酸标签的抗体(antibody–oligo conjugates),让蛋白质水平的定量也能通过测序来获取。
# 实验流程
抗体标记
- 每种待测抗体共轭一个短的 DNA 报告子序列(antibody barcode)。
细胞染色
- 将多重标记的抗体混合物与细胞(或细胞悬液)孵育,使抗体特异性结合对应表面蛋白。
单细胞分离与裂解
- 如同 scRNA-seq,包封单细胞并裂解,同时释放 mRNA 和抗体上的 DNA barcode。
共逆转录与扩增
- 同时逆转录 mRNA(带细胞条形码 + UMI)与抗体 DNA barcode。
文库构建与测序
- 分离两种 cDNA:一部分用于测序转录组,另一部分用于测序抗体验证条形码(常用定制引物扩增)。
数据融合
- 将每个细胞的基因表达数据与对应的抗体信号(epitope 数量)整合,得到 “RNA + 蛋白” 联合矩阵。
# 优势
- 同时获得转录组与蛋白组信息,提高细胞类型鉴别的精度。
- 蛋白质水平数据对低丰度或翻译后调控的靶点更敏感。
- 可以同时检测几十到上百种表面 / 胞内蛋白。
# 局限
- 抗体质量和特异性决定实验效果,需要严格的抗体验证。
- 抗体–oligo 的成本和制备复杂度高于普通 scRNA-seq 试剂。
- 通量受抗体组数和测序深度限制。
# 应用场景
- 免疫细胞谱系分型(T 细胞亚群、B 细胞亚群鉴定)
- 转录组与表型标志物联合分析
- 细胞分化过程中蛋白与 RNA 调控耦合研究
- 临床样本中多标志物联合表型剖析
# 3. Single-cell ATAC sequencing (scATAC-seq)
(Assay for Transposase-Accessible Chromatin)
# 基本原理
scATAC-seq 用于在单细胞水平绘制染色质开放性(chromatin accessibility)图谱,揭示基因调控元件(如启动子、增强子)在不同细胞类型或状态下的可及性差异。
# 实验流程
单细胞分离
- 类似 scRNA-seq,可用微流控芯片(10x Genomics Chromium)或微流体滴式系统。
Tn5 转座酶标记
- 在裂解后的细胞核中加入加载了测序接头的 Tn5 转座酶,它会 preferentially 插入到开放染色质区,使这些区域同时获得测序接头。
条形码融合
- 通过在微液滴或孔室中完成转座反应,使插入片段带上细胞条形码和 UMI。
扩增与测序
- 扩增插入了接头的 DNA 片段,构建文库并进行高通量测序。
数据处理与分析
- 根据条形码将读数分配到细胞,去除 PCR 重复后得到每个细胞在基因组各位点的可及性矩阵(细胞 × peaks)。
- 下游可做细胞聚类、TF motif 分析、基因调控网络推断等。
# 优势
- 直接捕获调控元件活性,补充转录组数据无法提供的调控层信息。
- 能鉴定特异细胞类型的关键增强子或 TF 結合位点。
- 与 scRNA-seq 联合(例如 Multiome)可同时获取转录组和染色质可及性数据。
# 局限
- 通量和数据稀疏性问题更严重,通常每个细胞只有几千到一万条有效片段。
- 峰(peak)调用和比对较复杂,对计算资源要求高。
- 需要更深的测序深度以覆盖低频开放区域。
# 应用场景
- 构建细胞类型特异的调控元件图谱
- 推断转录因子活性与基因调控网络
- 发育过程中染色质重塑动态研究
- 结合 GWAS 数据定位变异影响的调控元件
总结对比
| 特性 | scRNA-seq | CITE-seq | scATAC-seq |
|---|---|---|---|
| 主要测量层面 | mRNA 转录本 | mRNA + 表面 / 胞内蛋白 | 染色质开放性(调控元件可及性) |
| 条形码与 UMI | 都有 | 都有 | 都有 |
| 数据稀疏性 | 较高(dropout) | 较高(RNA 部分) + 蛋白部分相对低 sparsity | 非常高(开放片段稀疏) |
| 分辨信息 | 转录本丰度 | 转录本 + 表型蛋白 | 调控元件活性 |
| 典型通量 | 10⁴–10⁵ 细胞 | 10⁴ 细胞(视抗体数量与测序深度而定) | 10³–10⁴ 细胞 |
这三种技术各有侧重,常常结合使用以获得细胞多层次的分子图谱,帮助我们更全面地理解细胞状态与功能。
说实话,总感觉这段话有点牵强,真的和 perturbations 相关性这么大吗。
下面的一段说了一下大规模数据的影响,目前精确去预测网络的 interactions 还是受限于数据规模小的影响。Large-scale single-cell perturbation–response screens enable exploration of complex cellular behavior inaccessible in bulk measurements.
研究目的:Reliable analysis of increasingly large perturbation datasets requires efficient statistical tools to harness large numbers of cells and perturbations. 研究背景:The inherently high dimensionality of perturbation–response data complicates calculation of distances between perturbations, as does cell-to-cell variation and data sparsity. 本文基于各种 Multiple distance measures 方法中的 energy distance。
“Cell lines”(细胞系)是指在体外条件下(通常是培养皿或培养瓶中),从多细胞生物体取出的细胞经适当处理后,能够持续生长、分裂并保持特定特征的一类细胞群体。
Large perturbation screens are specifically designed to study a particular system, such as a cell line, under a set of perturbations of interest. 研究背景:“the field has accumulated a heterogeneous assortment of single-cell perturbation–response data with a wide range of different cell types, such as immortalized cell lines and induced pluripotent stem cell-derived models, and different perturbation technologies, including knockouts, activation, interference, base editing and prime editing”。所以 Computational methods to efficiently harmonize these different perturbation datasets are needed.
所以这个文章干了两件事,第一,提供了 scPerturb 去标准化数据集,第二,提供了 E-distance 作为衡量指标。
还是有点懵。
# Results
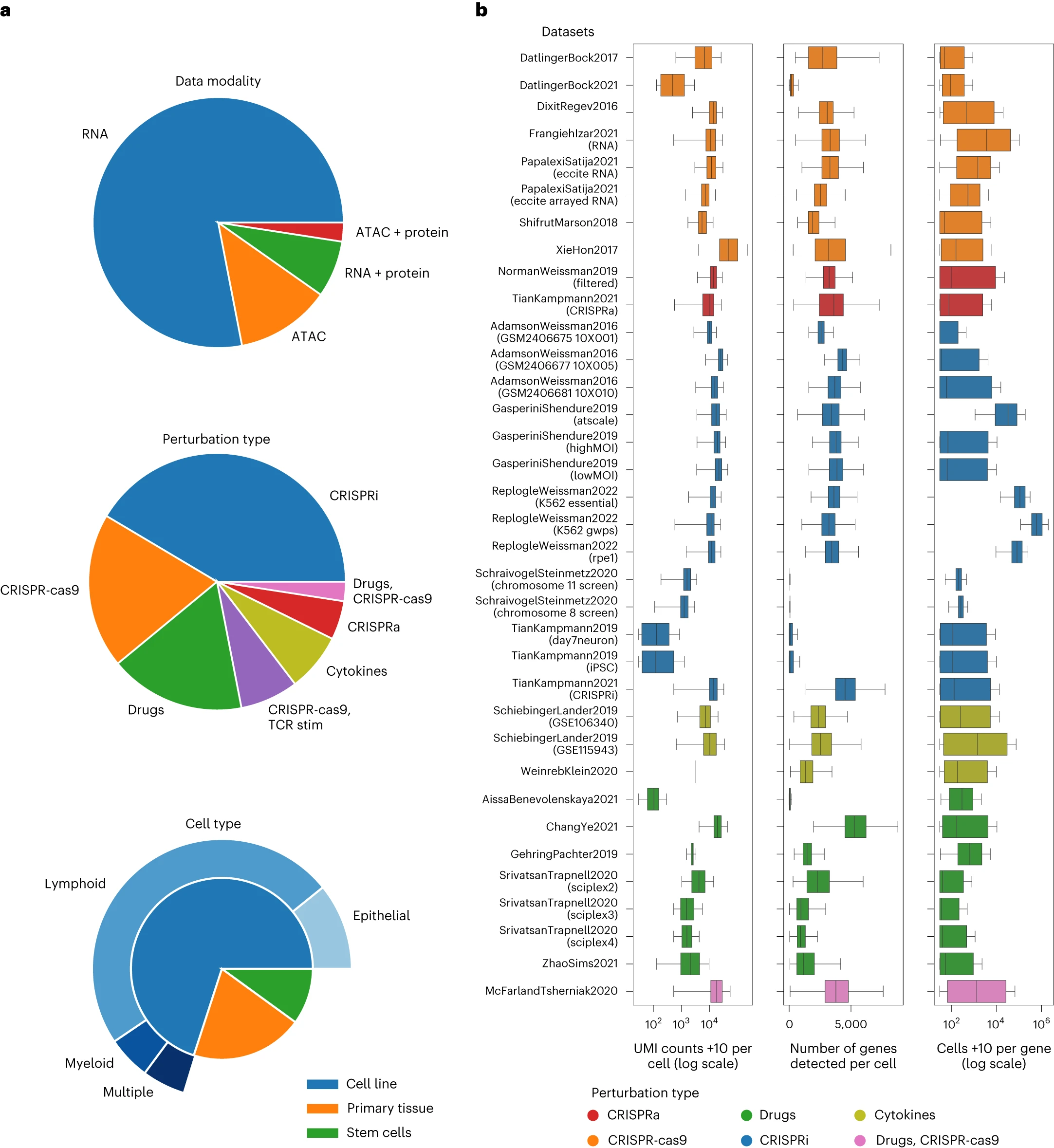
这里记一个结论:The average sequencing depth, that is, the mean number of reads per cell, in each study affects the number of low-expression genes observed. Increasing sequencing depth increases the UMI counts measured even for low-expression genes, reducing the uncertainty associated with zero counts.
“测序深度(sequencing depth)” 是指在测序实验中,每个位点或每个分子被测序的平均次数,它反映的是数据的丰富程度或覆盖程度。
这一大段挺难理解的,我拿 GPT 跑了一个解释:
这段话的核心在讲:不同数据集的测序质量指标差异很大,而测序深度对单细胞扰动实验的可解释性和分析结果有显著影响。
具体讲了以下几点:
# 🔍 1. 不同数据集的样本质量差异显著
数据集中细胞总数的多少主要受限于实验条件(例如样本处理能力、试剂成本、设备通量),尽管随着技术进步总量有增长趋势(见 Fig. 2b)。
常用的质量控制指标包括:
- 每个细胞的总 UMI 数(unique molecular identifier)
- 每个细胞检测到的 基因数量
👉 这些数值反映了测序的 “有效信息量”,在后续数据分析中用于筛选低质量细胞。
# 📊 2. 平均测序深度影响低表达基因的检测
平均测序深度 = 每个细胞平均测得的 reads 数量
测序越深,就越可能捕获到低表达的基因,UMI 数也越高,进而:
- 减少 “0 表达” 现象的误判(dropout)
- 提高低表达基因的可靠检测
这意味着:深度高不仅能检测更多基因,还能降低因低丰度而产生的假阴性
# 🧪 3. 测序深度 ≠ 数据质量
并非只要加深测序就能弥补实验缺陷:
- 如果在实验过程中 mRNA 已经降解了,reads 再多也无法 “复原” 丢失的转录本。
- 数据恢复的上限受限于样本质量,常用 ** 测序饱和度(sequencing saturation)** 来估计。
# ⚠️ 4. 这些质量差异会影响后续分析
不同扰动条件下,如果测序深度或数据质量不一致,可能会影响:
- 扰动间的差异是否能被识别(distinguishability)
- 聚类、差异表达、轨迹分析等下游计算的效果(downstream analysis performance)
这里介绍了 E-distance 这个概念,对于两组细胞,它描述了这两组细胞的分布的 distance。More precisely, it compares the mean pairwise distance of cells between two different perturbations to the mean pairwise distance of cells within each of the two distribution.
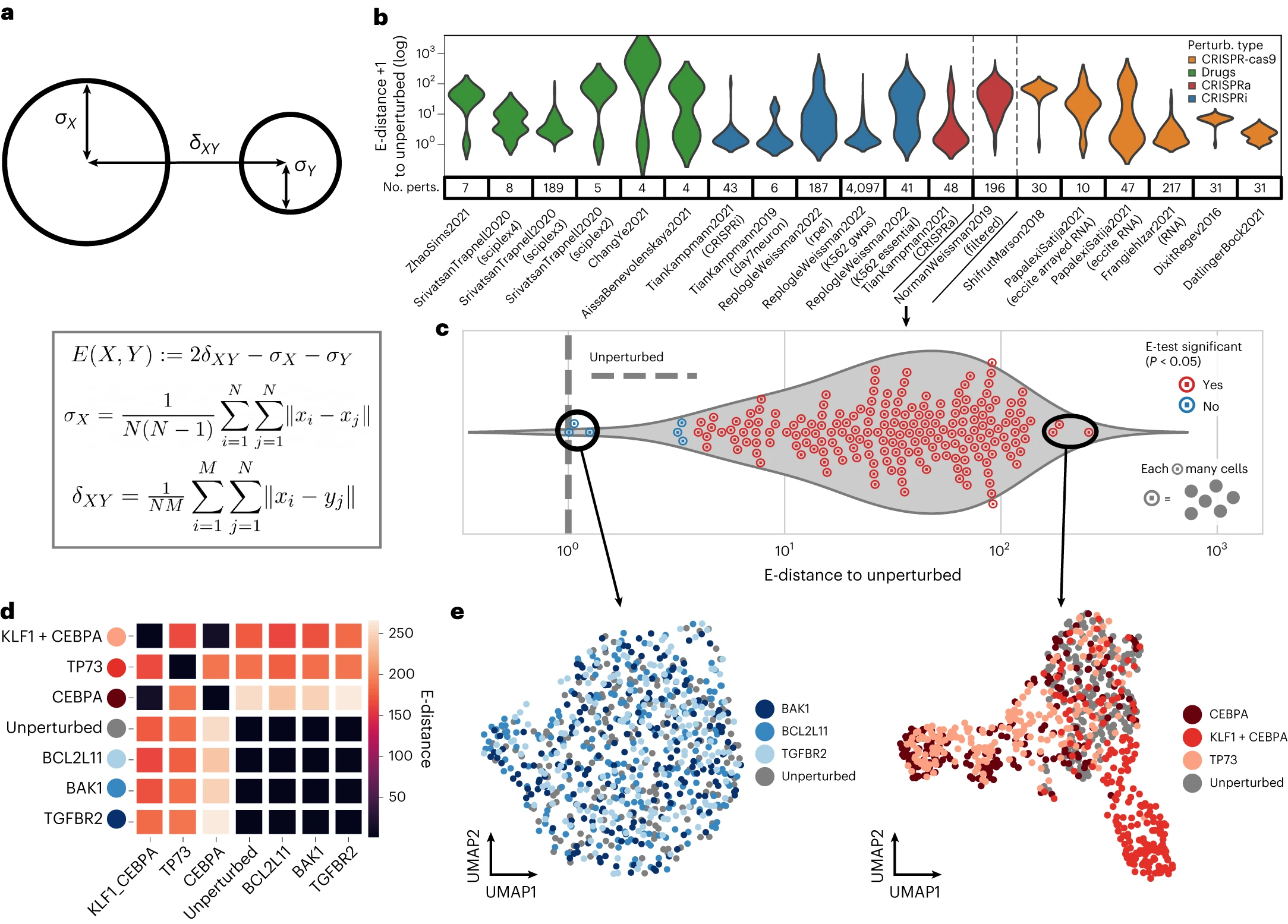
Fig (c) :This is in part caused by two-target perturbations using CRISPRa in that dataset: targeting the same gene with two single guides increases the chances of causing a considerable change in the transcript profile.
The E-distance can also be used to quantify similarity between different perturbations.
Fig (e): 观察 unperturbed 和其它的分布可以看出对于不同的 E-test 值可以明显区分出来。对于 CEBPA 和 KLF1+CEBPA 重叠的区域 is captured by a low E-distance:cells affected by these two perturbations are closer to each other than they are to unperturbed cells or to those from other perturbations.
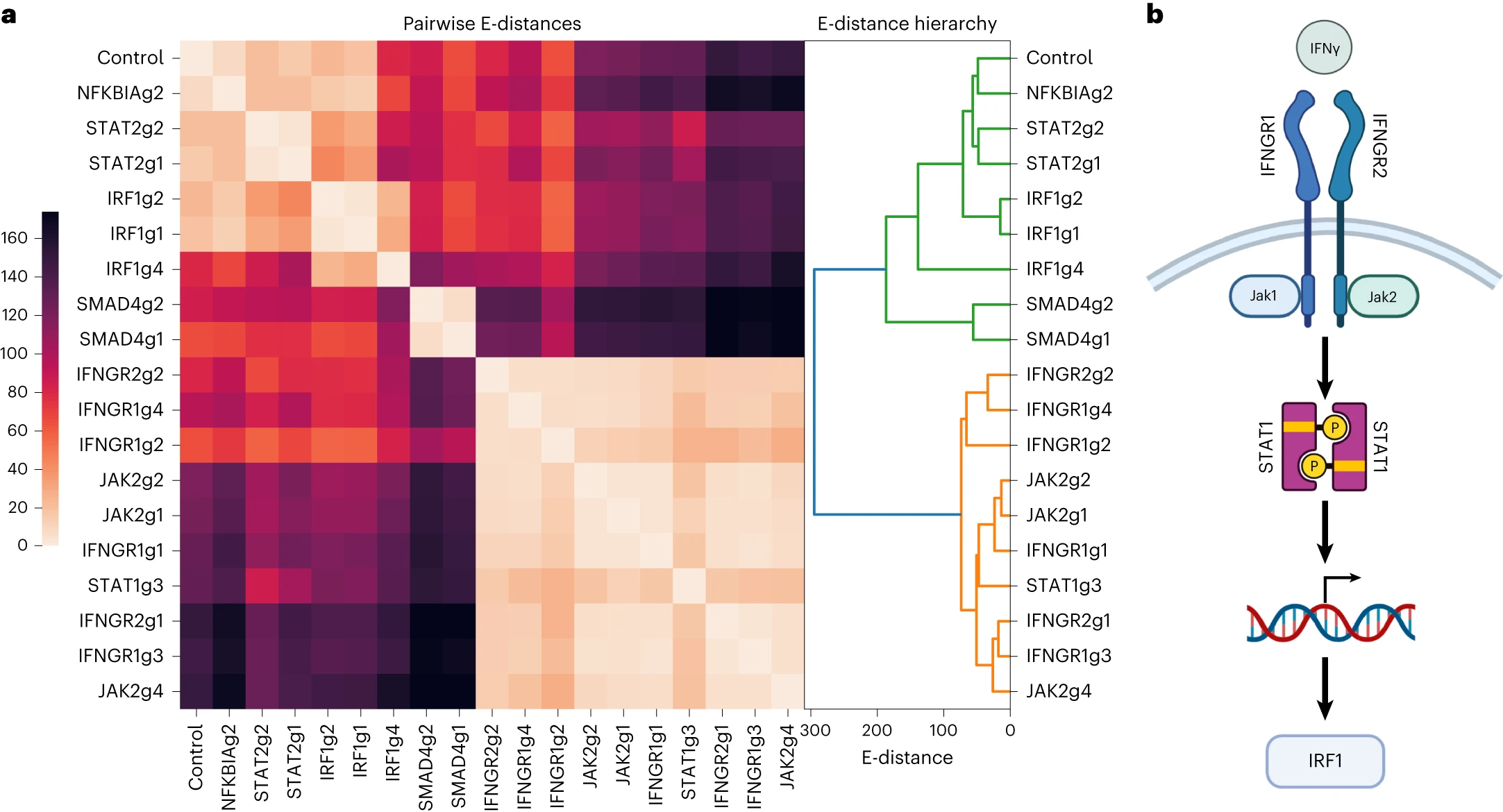
这段话主要在说明:如何利用 E-distance(能量距离)分析扰动数据集中不同基因扰动之间的相似性,并验证其在免疫信号通路中的生物学一致性。
# 📌 具体讲了什么?
分析背景与数据集来源
- 研究选用了一个关于免疫抑制检查点的扰动数据集(见 Fig. 4),该数据集使用 CRISPR-Cas9 对多个调控 PD-L1 表达 的基因进行扰动(参考文献 [9])。
方法:E-distance 计算扰动间的相似性
- 计算了所有基因扰动对之间的 两两 E-distance,即它们诱导的细胞状态差异有多大。
- 然后对这个距离矩阵进行了层次聚类(hierarchical clustering),发现出现了两个显著不同的扰动子群(clusters)。
观察与解释:形成一个具有相似表型的基因群
- 其中一个扰动群体(IFNGR1、IFNGR2、JAK2、STAT1)之间的 E-distance 较小,说明它们产生了相似的细胞转录组响应。
- 而它们与 “未扰动对照组” 之间的 E-distance 较大,说明这些扰动引起了显著变化。
生物学一致性验证
这组基因(IFNGR1, IFNGR2, JAK2, STAT1)正好都位于 IFNγ(干扰素 -γ)信号通路中,属于同一个上游 / 下游调控链条:
- 它们都在 IFNγ → IRF1 → PD-L1 表达 的通路中起作用(参考文献 [50])。
因此扰动这些基因会引发相似的表型改变,是功能一致性的体现。
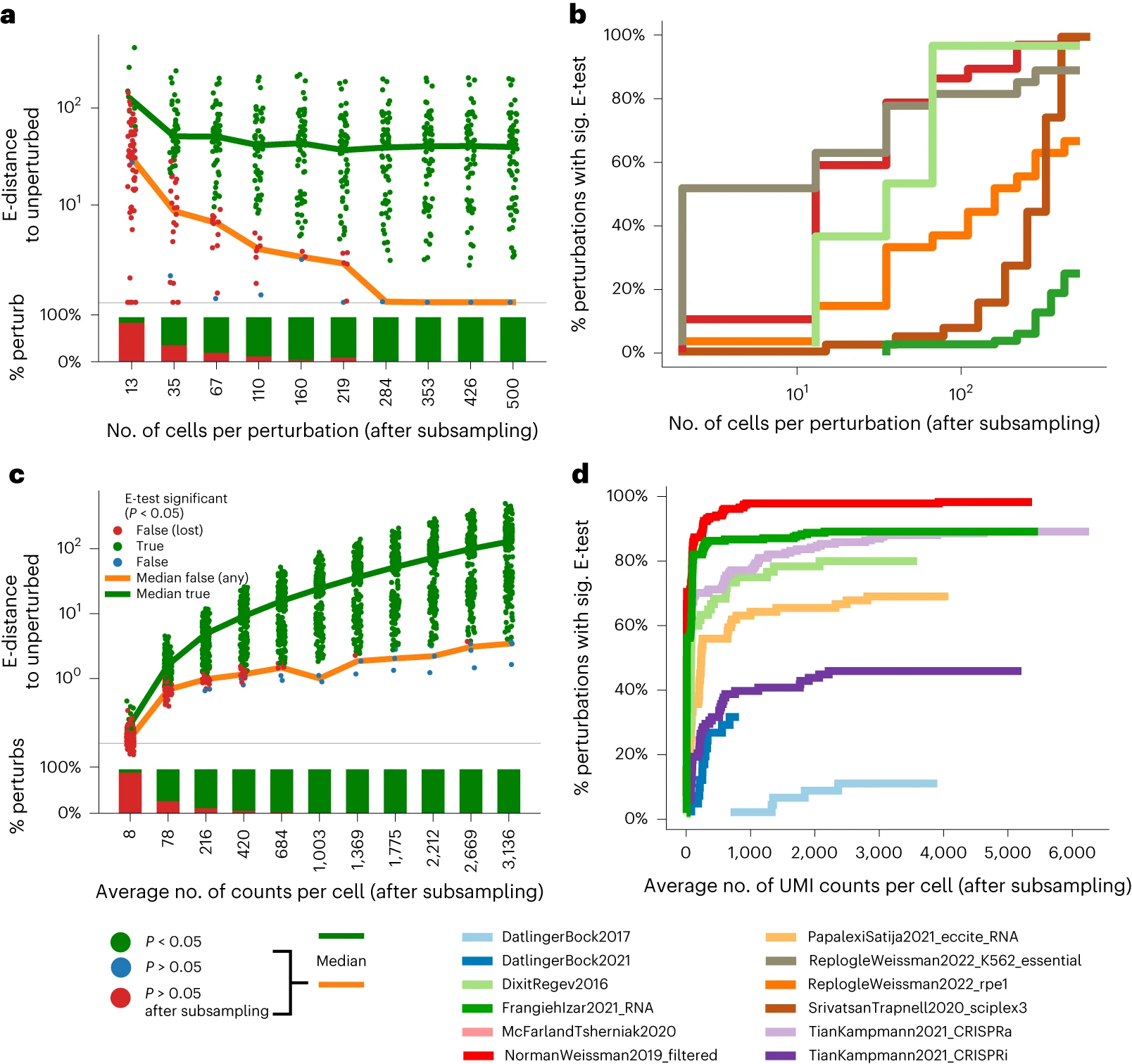
这段话讲的是:作者通过子采样(subsampling)实验系统评估了 E-distance 和 E-test 对样本数量变化的敏感性和鲁棒性(稳定性),并给出合理使用它们的建议。
# 📌 具体讲了什么?
# ✅ 1. 研究目的
- 使用 scPerturb 提供的统一注释的单细胞扰动数据集,评估 E-distance(用于量化扰动差异)和 E-test(用于显著性检验) 对实验设计和数据处理参数的鲁棒性(尤其是细胞数目的影响)。
# ✅ 2. 实验方法:人为减少细胞数进行测试
- 人为下采样每个扰动条件中的细胞数,模拟 “小样本” 场景,观察这时 E-distance 和 E-test 分数如何变化。
# ✅ 3. 偏差修正(bias correction)
- 提出了一种 E-distance 的偏差修正方法,以提高在小样本条件下的准确性(详见补充材料第 1 节)。
# ✅ 4. 发现 1:即使修正后,E-distance 在细胞数减少时仍上升
即使使用了偏差修正,在每个扰动下的细胞数减少时,E-distance 仍会人为变大,这说明:
- 计算 E-distance 前需要对每个扰动的细胞数进行标准化(例如统一抽取相同数量的细胞),否则可能引入比较偏差。
- 原因在于:PCA 在小样本情况下不能很好表示高维数据结构,从而扭曲了距离估计。
# ✅ 5. 发现 2:显著性检验结果随着样本数下降而自然减少
- 当细胞数下降时,能被识别为显著扰动的基因数量减少,这是符合预期的:样本越少,统计功效越低。
- 同时观察到,并非所有数据集在 “全部细胞数” 时都达到了扰动检测的饱和状态(即:进一步增加细胞数可能还会发现新扰动)。
# ✅ 6. 影响因素:扰动强度与数据异质性
检测 “饱和点”(saturation point)取决于:
- 扰动的效应强度(effect size):强效扰动易被发现,弱效扰动需要更多细胞。
- 数据集的异质性(heterogeneity):如果细胞彼此非常相似,则较少样本就足以捕捉扰动的平均效应。
这段话主要讲的是:评估单位细胞内的测序深度(即 UMI 数量)对扰动分析结果的影响,并提出实际实验设计的参考建议。
# 📌 具体内容逐句解析:
# ✅ 1. UMI 数越多,E-distance 越大
“We subset the number of UMIs per cell, finding that E-distance increases as the UMI count per cell increases (Fig. 5c).”
作者控制变量,只改变每个细胞的 UMI 数(模拟不同测序深度):
- 结果发现:随着每个细胞的 UMI 数增加,E-distance(能量距离)也增加。
- 原因可能是:高 UMI 会暴露出更多细微的扰动效应,使得扰动组与对照组在表达空间中的距离拉大。
# ✅ 2. 但 E-test 的显著性结果在~500 UMI 就饱和了
“The number of significant perturbations under the E-test, however, saturates at around 500 UMIs per cell...”
虽然 E-distance 随深度增加而变化,但用于判断扰动是否显著的 E-test(统计检验) 在 UMI 达到 500 左右时,显著性结果就趋于稳定(饱和):
- 也就是说:即使 UMI 较少,统计显著的扰动大多数依然能被检测到。
# ✅ 3. 说明 E-test 更稳定、更实用
“The stability of E-test results... demonstrates the necessity of the E-test... as the appropriate statistical measure...”
E-test 比 E-distance 更稳健、不那么依赖测序深度,因为:
- 它使用了 随机化对照(permutation control),可以抵消一部分数据量波动带来的影响。
- 所以在实际分析中,E-test 是更适合用来判断扰动是否有效的统计工具。
# ✅ 4. 实验设计建议(干货!)
“we suggest at least 200–500 cells per perturbation and an average of 1,000 UMIs per cell...”
综合分析后,作者给出一个推荐标准,用于指导单细胞扰动实验设计:
- 每个扰动条件下建议至少测序 200–500 个细胞
- 每个细胞建议测出平均 1,000 个 UMI
- 这两个条件基本能保证对多数 “显著扰动” 的稳定检测。
# ✅ 5. 但最佳测序深度还要看任务
“The optimal lower bounds... depend on downstream specific modeling tasks...”
注意:这个 “推荐标准” 是用于 “检测扰动是否显著” 的一般性指导;
- 若下游任务更复杂(如预测建模、轨迹分析等),所需的测序深度和细胞数可能更高。
# Methods
终于到方法的部分了。
# scATAC-seq
scATAC-seq is a biomolecular technique to assess chromatin accessibility in single cells.
这段话主要讲的是:作者如何处理 scATAC-seq 数据以提取标准化的特征,用于分析单细胞染色质可及性信息。
# 📌 逐句解释如下:
# ✅ 1. 技术背景
“scATAC-seq is a biomolecular technique to assess chromatin accessibility in single cells.”
- scATAC-seq 是一种单细胞分辨率的技术,用于检测染色质的开放程度,即哪些 DNA 区域对转录因子等蛋白 “可及”(accessible)。
# ✅ 2. 数据输入格式
“The starting point of our data processing pipeline is BED-like tabular fragment files...”
- 数据处理流程的起点是类似 BED 格式的片段文件(tab-delimited),每一行代表一个被测序捕获到的 ATAC 片段(fragment),包含它所在的基因组位置和对应的细胞条形码(cell barcode)。
# ✅ 3. 数据处理目标
“The goal... is to extract standardized features from this information.”
- 目标是从这些原始片段信息中提取出一系列标准化的、可用于分析的特征。
# ✅ 4. 提取的五类特征如下:
LSI Embeddings(低维嵌入)
“embeddings derived from latent-semantic-indexing (LSI) with 30 dimensions...”
- 使用 潜在语义索引(LSI) 方法将高维稀疏的 ATAC-seq 数据降维成每个细胞一个 30 维向量,适合稀疏性强的单细胞 ATAC 数据。
Gene Scores(基因开放性得分)
“gene scores that measure the chromatin accessibility around each gene...”
- 每个细胞对每个基因,计算其 转录起始位点(TSS)邻近区域的开放程度加权和,即基因周围的染色质可及性,距离越远的片段权重越低。
Peak–Barcode Matrix(峰 × 细胞矩阵)
“a peak–barcode matrix that quantifies the chromatin accessibility at consensus peaks...”
- 构建一个 细胞 × 共识峰(peaks) 的矩阵,用于表示各细胞在特定基因组区域(peaks)上的染色质开放强度。
chromVAR Scores(转录因子活性分数)
“chromVAR scores, which quantify the activity of a set of transcription factors...”
- 利用 chromVAR 工具 计算每个细胞中一组转录因子对应的 DNA footprint 区域的活性变化,用来反映转录因子的功能活性水平。
Marker Peaks(扰动标志峰)
“marker-peaks per perturbation target, which quantifies the differential regulation...”
- 对每种扰动条件,识别出差异化活跃的高变峰(highly variable peaks),作为该扰动的 “标志性染色质调控区域”。
# scRNA-seq
这段话讲的是:作者如何处理和标准化各个原始单细胞扰动数据集,以便进行统一分析。
下面是逐句解释:
# 📌 1. 起始数据来源与处理方式
“Analysis was initiated using unfiltered, unnormalized cell-by-gene matrices as provided by source papers.”
- 分析从原始文献提供的 cell-by-gene 矩阵(即细胞 × 基因的表达矩阵)开始;
- 使用的是未经过滤或归一化的原始版本,确保保留最大的信息量,便于统一处理。
# 📌 2. 针对缺少处理后的数据集
“For one dataset, preprocessed cell-by-gene matrices were unavailable...”
- 有一个数据集并未公开处理好的表达矩阵;
- 作者就按照原论文中描述的流程,使用原作者提供的代码自行进行预处理。
# 📌 3. 细胞条形码(barcode)处理
“For datasets with cell barcodes, barcode assignments for cells were taken from the original paper when available...”
对于包含细胞条形码(即识别每个细胞身份)的数据集:
- 如果原论文提供了 barcode 到细胞的映射,作者就直接采用原始提供的数据;
- 如果没提供,就按原论文方法部分自行分配 barcode(详见方法部分)。
# 📌 4. 处理同一细胞含多个引导序列(guide)的情况
“If multiple guides were assigned to the same cell...”
如果一个细胞中检测到多个干预(例如多个 CRISPR guide RNA):
- 作者将这些 guide 按照计数从高到低的顺序排列,记录在最终的分析对象中;
- 这样可以保留 “主要扰动” 的信息,同时也不完全丢失次要扰动。
# 📌 5. 代码开放性
“The code used to process each individual dataset... is available in our code repository.”
- 作者将所有数据处理过程的代码(包括 barcode 分配)公开在他们的代码仓库中,确保透明和可复现。
# E-distance
Let and be samples from two distributions , corresponding to two sets of and cells, respectively.
We define :
\begin{equation} \delta_{XY} = \frac{1}{NM}\sum^N_{i=1}\sum^M_{j=1}{||x_i-y_j||} \end{equation} \begin{equation} \sigma_X = \frac{1}{N^2}\sum^N_{i=1}\sum^M_{j=1}{||x_i-y_j||} \end{equation}and defined accordingly.
Intuitively, is the mean distance between cells from the two distributions, while describes the mean distance between a cell from to another cell from . The energy distance between and is defined as:
\begin{equation} E(X,Y):=2\delta_{XY}-\sigma_X-\sigma_Y \end{equation}For the bias-corrected energy distance, we define
\begin{equation} \sigma_X = \frac{1}{N(N-1)}\sum^N_{i=1}\sum^M_{j=1}{||x_i-y_j||} \end{equation}这比原来的 $\frac1}{N,因为:
原公式包含了自己和自己的距离
而修正版本只考虑不同的样本对,避免了系统性低估组内变异性;
修正后更适用于小样本(如每个扰动只有几十或几百个细胞)的情况。
# E-test calculation
E 检验(E-test)作为一种 Monte Carlo 置换检验,以能量距离(E-distance)作为检验统计量进行实施。对于每个数据集中的每一个扰动条件,我们将该扰动组的细胞与未扰动对照组的细胞合并,然后随机打乱扰动标签,将细胞重新分为两组,并计算这两组之间的 E-distance。我们将这一过程重复 10,000 次。在这 10,000 次中,打乱标签后计算得到的 E-distance 大于原始未打乱标签所得到的 E-distance 的次数,除以 10,000,即得到了对应的 P 值,该值表示一个 单边检验(one-sided test) 的显著性水平。我们为本资源中几乎所有的数据集都报告了该检验的结果(见补充表 3)。
对于每个数据集中的多重假设检验,我们采用 Holm–Sidak 方法 进行多重比较校正。