MENDER: fast and scalable tissue structur identification in spatial omics data.
这次笔记尝试一下只记录我认为重要的地方,就不按部就班的来了,但总体还是分为文章 - 代码的结构
# 摘要
总体概括以下这个 MENDER (Multi-range cEll conNtext DEciphereR) 干了什么:
- offers substantial improvements over modern complex models while automatically aligning labels across slices, despite using much less running time than the second-fastest.
- MENDER's identification power allows the uncovering of previously overlooked spatial domains that exhibit strong association with brain aging.
- MENDER's sclability makes it freely appliable on a million-level brain spatial stlas.
- MENDER's discriminative power enables the differentiation of breast cancer patiant subtypes obsured by single-cell analysis
# Introduction
In a typical SRSC dataset, the spatial coordinates and gene-expression profiles of each cell are measured. Such datat representation naturally forms a spatial graph with cells as nodes and gene expression as node attributes, which motivated the two major modeling paradigms in this field:
- Graph Neural Network(GNN)
- GNN-based methods introduced dedicated neural modules, loss functions, and network architectures.
- Bayesian Network(BN)
- BN-based methods extend additional hideen variables, varianle dependencies, and specified priors.
MENDER has 3 highlighted points:
- multi-slice spatial domain identification that challenges many advanced methods.
- scalability to million-level datasets
- improved running time efficiency without the need of GPU
# Results
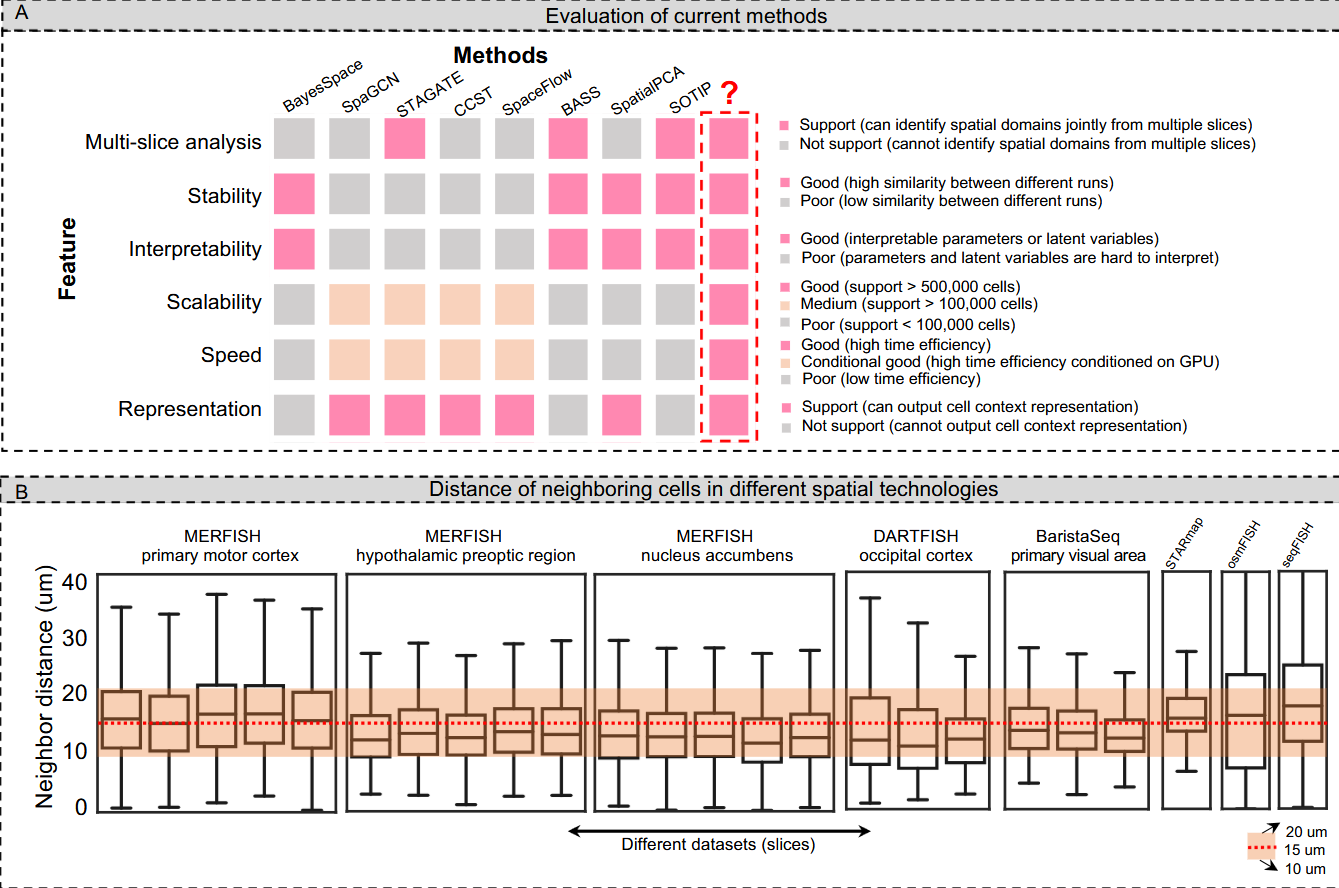
All GNN-based methods have better scalability and speed than BN-based methods and they can also output the context representations for cells.
The common limitations of GNN-based methods are the lack of stability and interpretability inherited from general deep-learning models.
BN-based methods, on the contrary, have better output stability and interpretability than GNN-based methods since they are generally built on well-defined probabilistic variable dependencies.
But they cannot guarantee good scalability to large datasets with short running time, and generally don’t output the cell context representations
MENDER 的 Overview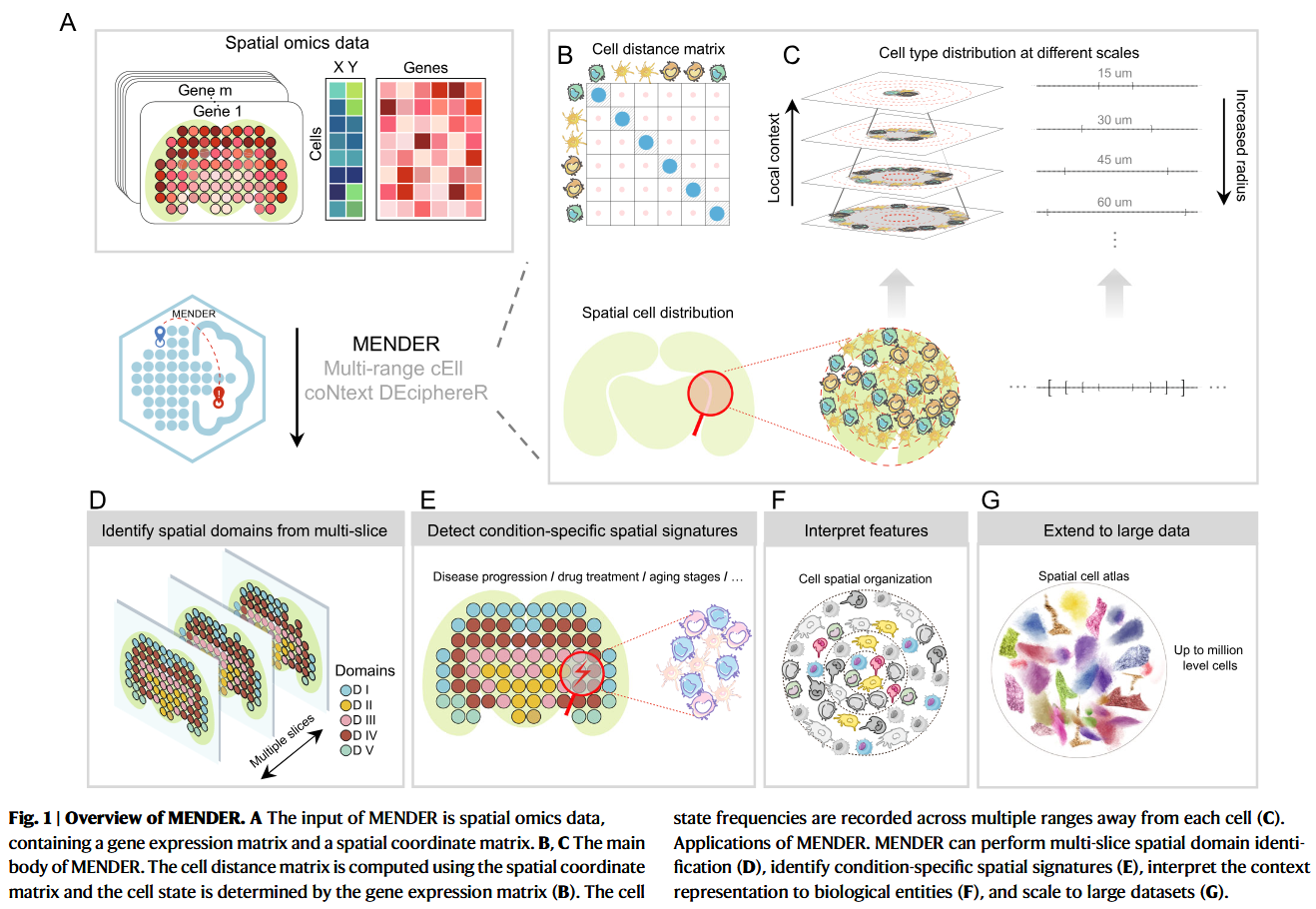
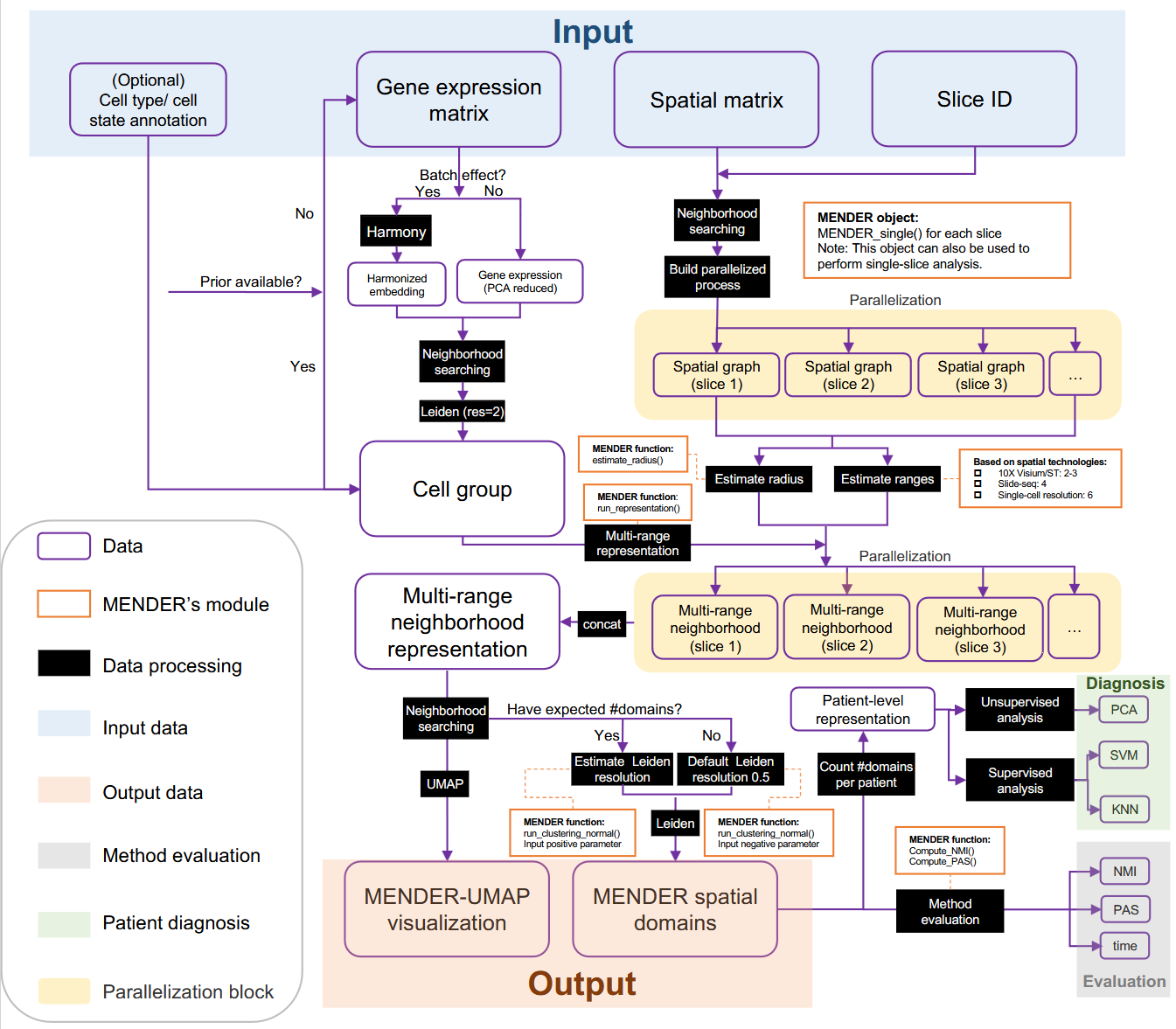
这玩意不结合数学公式来看确实也看不懂,我这里直接把 Methods 里面的东西弄过来
# 输入
G genes, M cells from S slices
The "Gene expression matrix"(Nrows Gcolumns)
The "Spatial matrix (Nrows 2 columns, for 2D data, 3 columns for 3D data)
The slice ID identifier(a vector of length N)
# Cell group compution
说白了就是用 Leiden 给 gene expression 数据聚类,得到 Cell group,分两种情况(batch effect)
# Multi-range neighborhood Representation Computation
根据上面的操作,现在手上的数据有 "Spatial matrix", "Silce ID", and "Cell Group"
首先,为了放着不同 slice 的细胞成为 neighbors,先将它们按照 slice 进行 parallelization 操作.
For each slice, around every cell, S ranges of spatial neighborhoods are created, forming S ring areas around the central cell (the radius is set to 15um by default). The cell index located within each ring area is recorded for each central cell.
这里的 ranges 指的是有多少个环,也就是 radius 扩大多少次。
Formally, suppose the total number of cells across all input slices is N, the first step partitioned all cells into C distinct cell states.
这里的 C distinct cell states 应该指的是 Cell Group 里面聚类出来的不同细胞类型
noted as ,
The cell state of the i-th cell is noted as ,.
The origin slice of the i-th cell is noted as ,.
The spatial coordinate of the i-th cell is nored as ,
The number of ranges if set to . The radius is set to .
Then the multi-range neighborhood representation matrix,, in which the i-th row is the context-aware representation of the i-th cell.
这里的 M 指的是满足这个条件的数量, 表示数量的计算。
# 总结一下
说白了这玩意还是把基因的信息和 spatial 的基因结合并凸显它们的特征然后利用得到的信息进行聚类,只不过把范围扩大到了多 slice 的情况。
# 评价环节
又到了喜闻乐见的吹水环节,这里就把我认为比较有代表性的图片粘贴上来
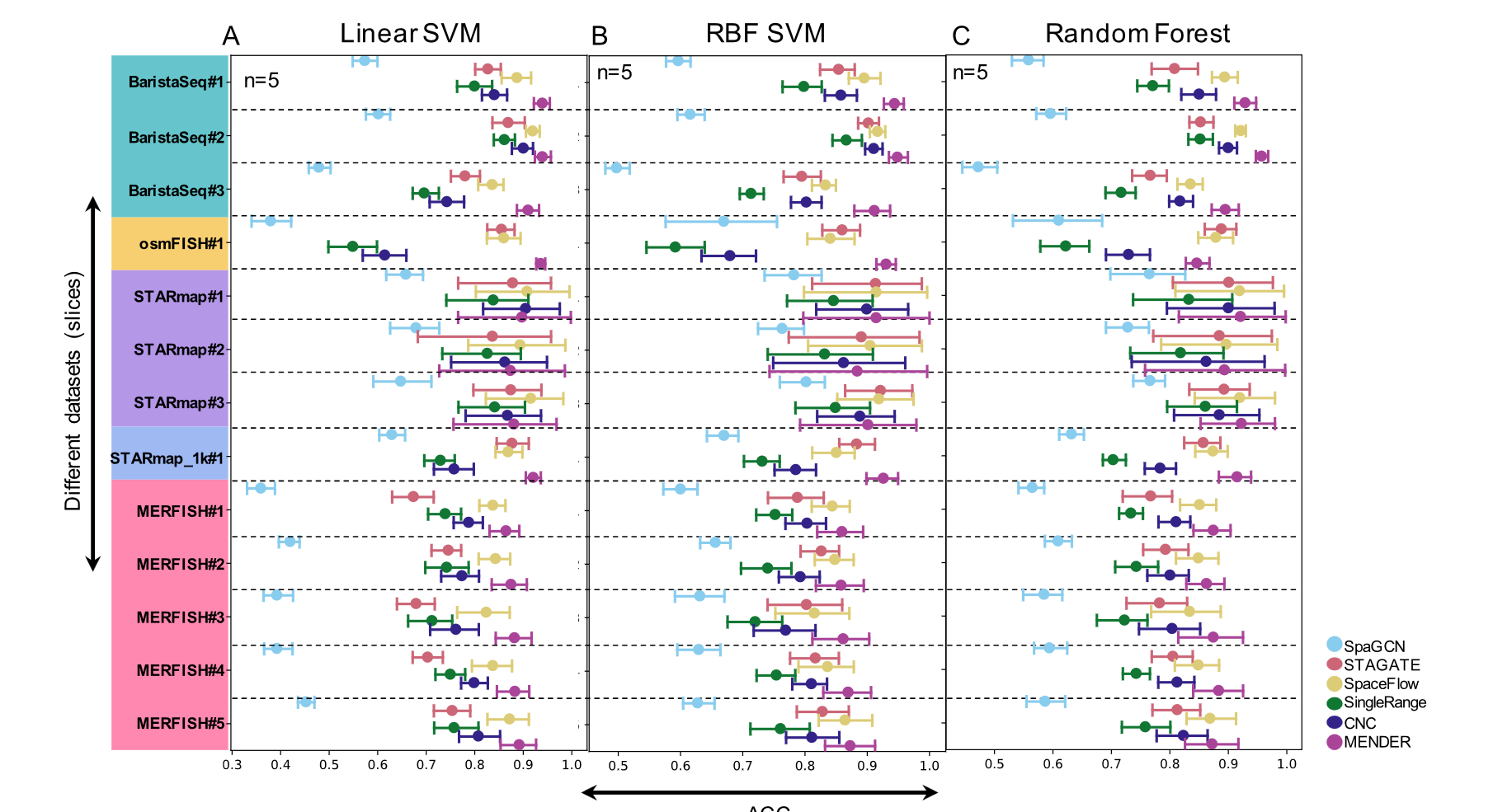

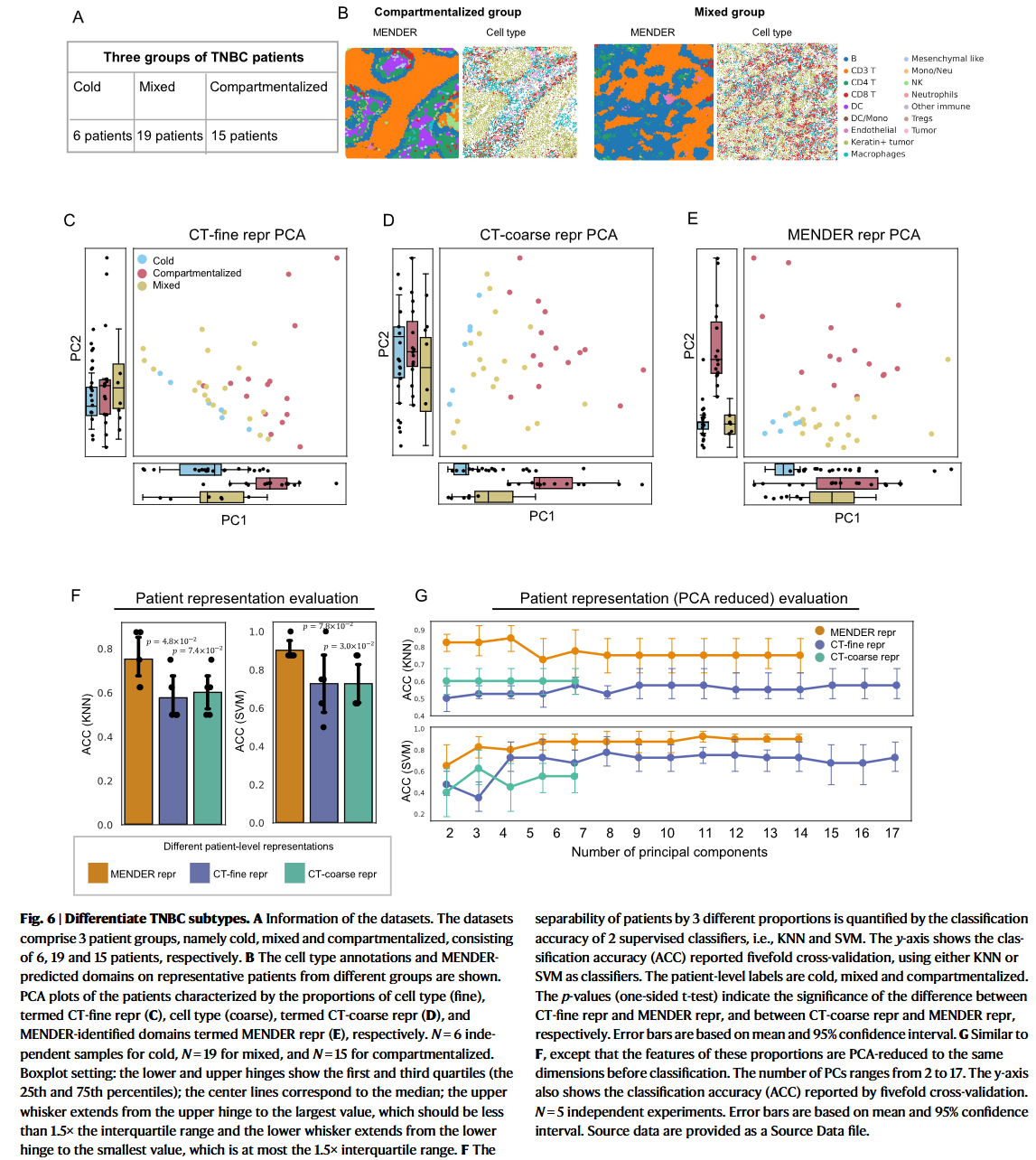
The original publication supplied both fine and coarse cell classifications.
we define patient representation using MENDER domain proportion as "MENDER repr".
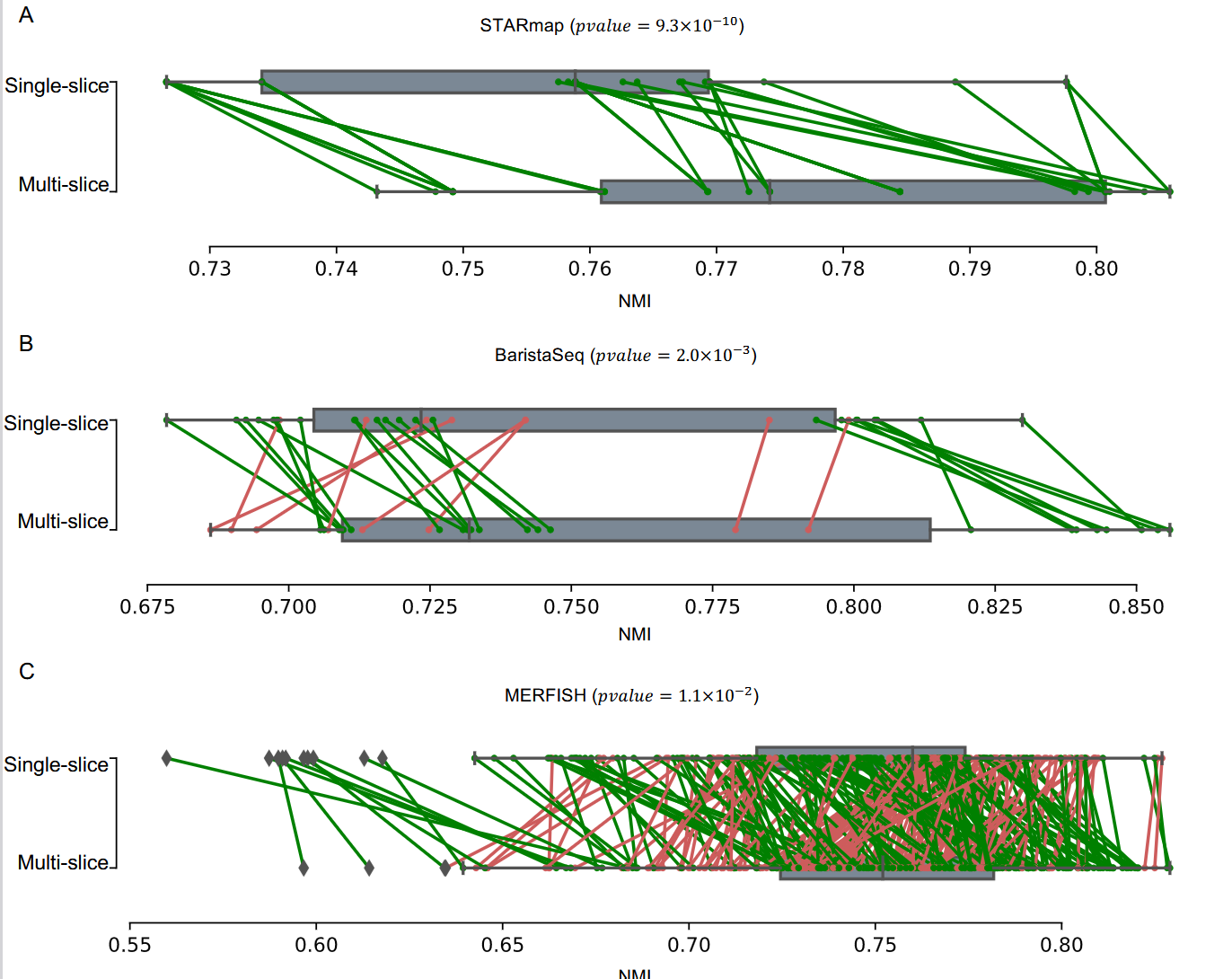
这里的每一个点指的是 S slice replicated runs
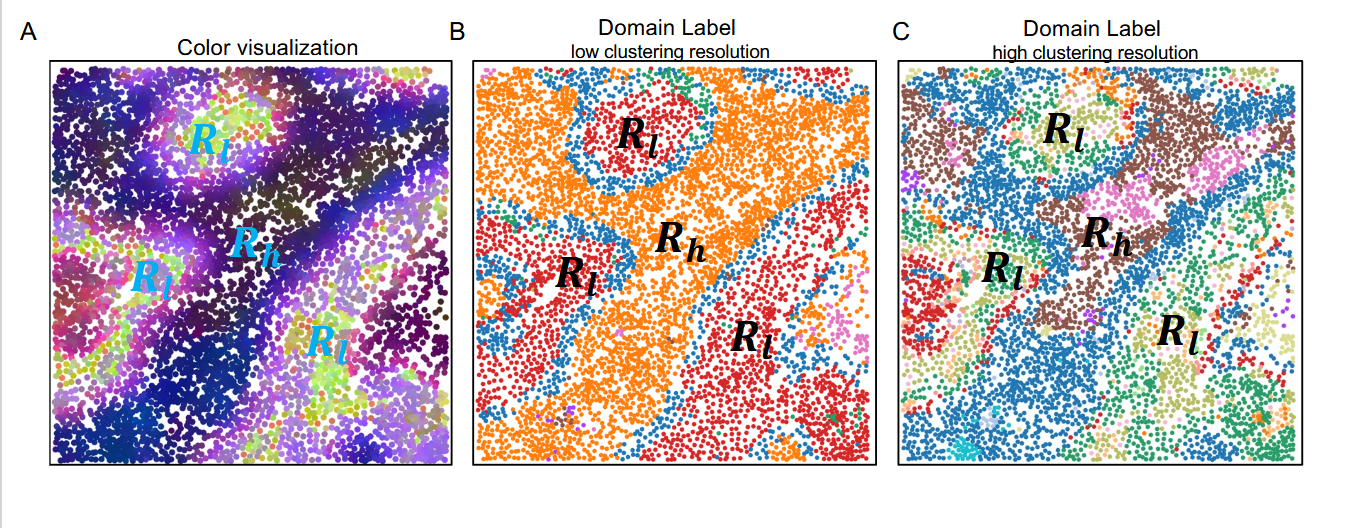
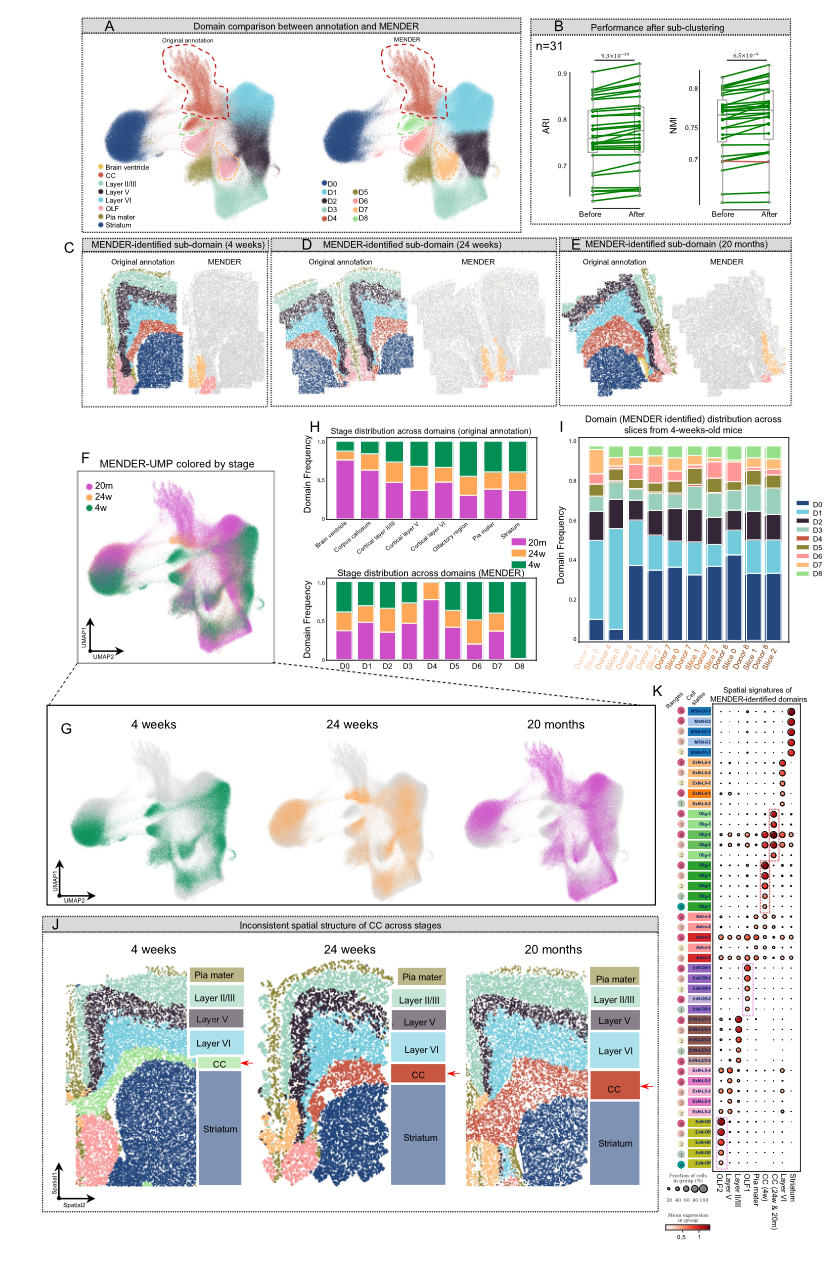
这张图片表示了对于同一个 spatial domain 也能体现出细胞 context 的 variation。对于后两个图片是基于不同的分辨率得到的。

这是对于图片的解释
# Discussions
这篇文章的 discussion 写的确实挺不错的,有些观点可以记录一下
There are primarily two factors that can influence the determination of spatial domain labels:
- The first factor is cellular context because MENDER relies on the representation of cellular context tot determine spatial domain labels.
- The second factor is the Leiden clustering resolution.
There were two folds of analytical contributions:
- we identified consistent nerghborhood statistics across different spatial technologies in different tissue systems,
- we found that simple cellular context analysis might have improved performance compared to state-of-art complex models in both supervised and unsupervised settings.
# Code
没什么好写的,就是把上面的内容给复现了罢了