# 单词表
单词表
scope: 范围
evaluate: 评价
scenario: 设想的情况
multiplex: 多路运输
pose: 构成
substantial: 可观的
annotation: 注释
conquer: 征服
extant: 尚存的
extensively: 全面的
laminar: 薄片 / 层型
NMI: normalized mutual information
incentivize: 激励
enccompass: 包含
silhouette: 轮廓
ASW: average silhouette width
PAS: perccentage of abnormal spots
adjacency: 邻接物
quantutative: 定量的
necessitate: 使・・・成为必要
cutting-edge: 简短
multiomic: 多组学
predominantly: 全面的
deconvolution: 去卷积
imputation: 归咎
modularity: 模块化
histological: 组织学
autoencoder: 自编码
facilitate: 促进
contingency: 应急事件
contingency tabble: 列联表
spectrometry: 质谱
manifold: 多种多样的
UMAP: uniform manifold approximation and projection
concatenate: 把・・・练习起来
batch effect: 批次效应
unadorned: 未修饰的
HOM: the homogeneity score
COM: the completeness score
# 文章概括
这篇文章就不摘要观点了,基本都是思路和算法,后面会详尽的列出算法和实现的 python 代码,大致是实现 SpaGCN (HE) 的代码
这篇文章主要是针对 13 种对于在 34 个 SRT (spatially resolved transcriptomics) 的数据的计算方法做了 benchmark study,并针对 accuracy,sptial continuity,marker genes detection,scalability,and robustness 做了评估。针对各个方法的特点提出了自己的选择建议。并在最后提出了自己针对大规模数据的 Method。

实验发现没有一种方法是适用于所有的数据的
选择方法和数据的网站
接着作者检测了现有方法的局限性,利用的数据是 (additional 22 data containing small and non continuous tissue domains, and during multislice analysis on another large-scale dataset containing 31 tissue slices)
作者提出解决局限性的策略是 'divide and conquer' strategy
最后,作者检测了鲁棒性和其它因素 (gene expression matrix sparsity, spatial resolution, the number of genes and the level of noise), 也检测了 pre- and postpocessing steps on diffenrent methods' performance.
As these data(10x Visium, stereo-Seq, BaristaSeq, MERFISH, osmFISH, STARmap and STARmap*) were pproduced using distinct spatial technologies, they exhibit different data characteristics and collectively span a wide range of potential spatiial transcriptomics data types, which allows users of different spatial technologies to benefit from study
- nonspatial methods
Louvain
Leiden - sptial methods
SpaGCN
BayesSpace
stLearn
SEDR
CCST
SCAN-IT
STAGATE
SpaceFlow
conST
BASS
GraphST
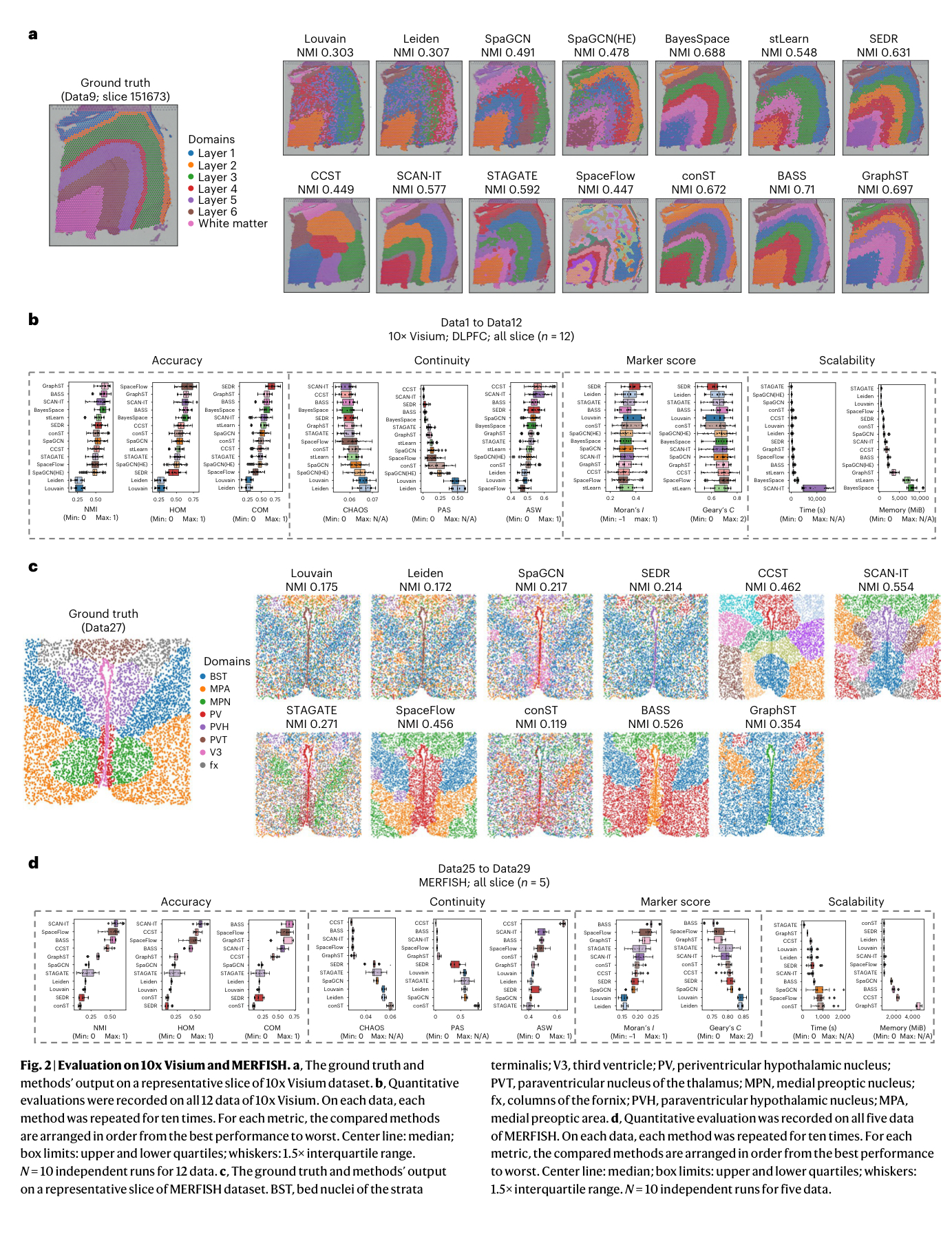
Benchmarking analysis on 10x Visium dataset
VS
Benchmarking analysis on MERFISH dataset
对于 Comprehensive evaluation:
- Accuracy
NMI
HOM
COM - Continuity:
CHAOS
ASW
PAS - The quality of domain-specific marker genes
Moran's I
Geary's C - Scalability
Time
Memory
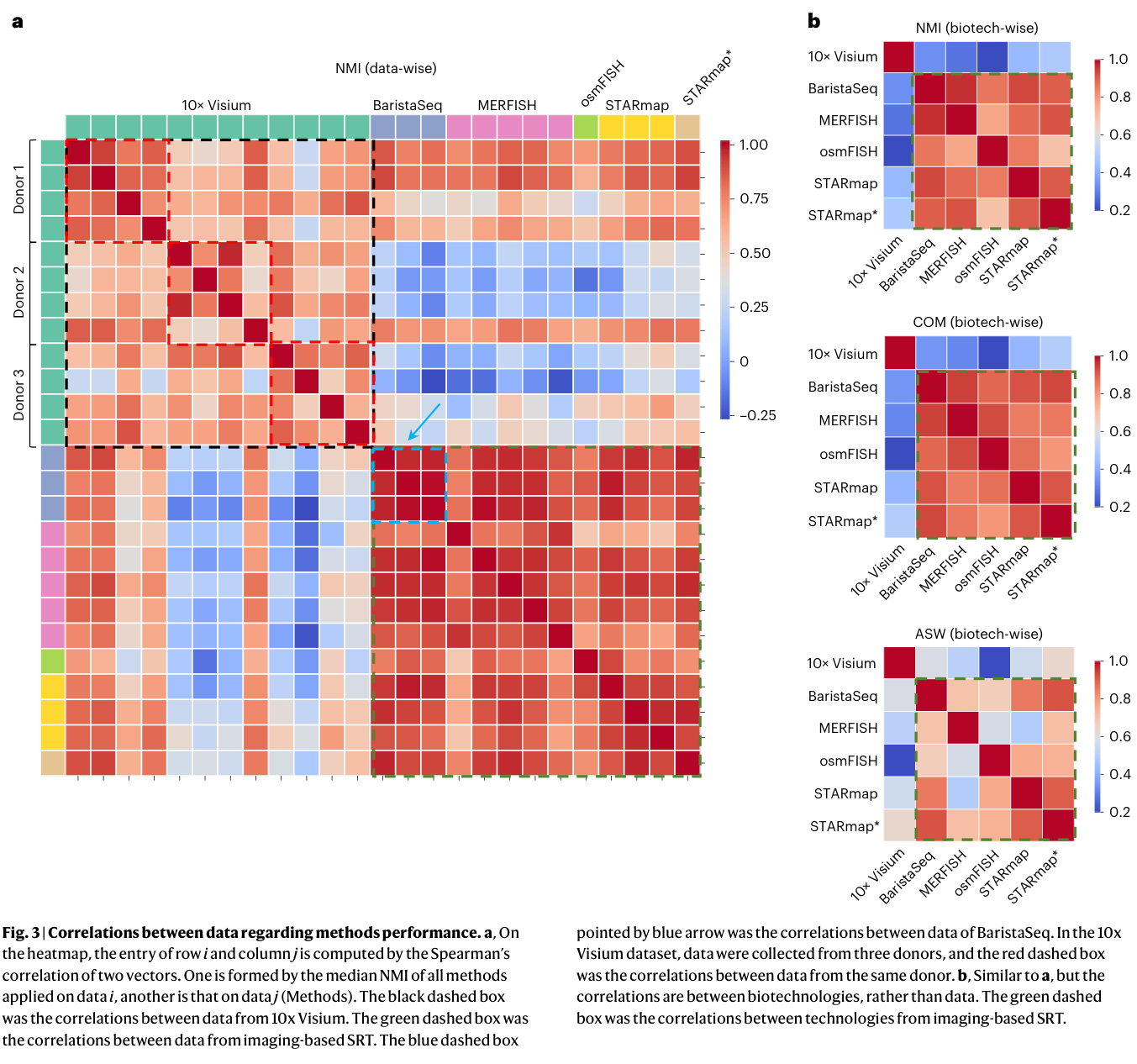
Through the analysis of methods performance across different data, we have observed interesting correlation patterns as determined by the Spearman correlation coefficient, underlining the relative ordering of their performance (Fig. 3a, the underlying metrics values can be found in Extended Data Fig. 2). First, we found that, within the same spatial technology, a strong Spearman correlation emerged when mul- tiple data samples originated from the same individual and shared identical tissue structure. A notable example of this is the high consist- ency in the relative performance of all methods applied on three BaristaSeq datasets (Fig. 3a, blue box).
Second, when tissue structure is controlled, methods performance may be influenced by the use of different technologies. Evidence for this is seen when comparing the performance of methods on the 10x Visium dataset with that of the BaristaSeq dataset. Sharing similar tissue structures (brain cortex region, Supplementary Table 1), the two datasets, generated by differ- ent spatial technologies, yielded different performance outcomes (Spearman’s correlation is 0.33, Fig. 3b).
Third, when spatial technology is controlled, the origin of the data samples (that is, different donors) can also influence method performance. An example is on the 10x Visium dataset, the significantly higher correlations (Supplementary Fig. 9, P = 4.65 × 10−4) when applying methods on data from the same donor than from the different donors (correlation values from the same donor are highlighted in red box in Fig. 3a)
- the performance of all methods bu partitioning the datasets into two groups:
10x Visium
imaging-based datasets(MERFISH, osmFISH, BaristaSeq, STARmap, STARmap*)
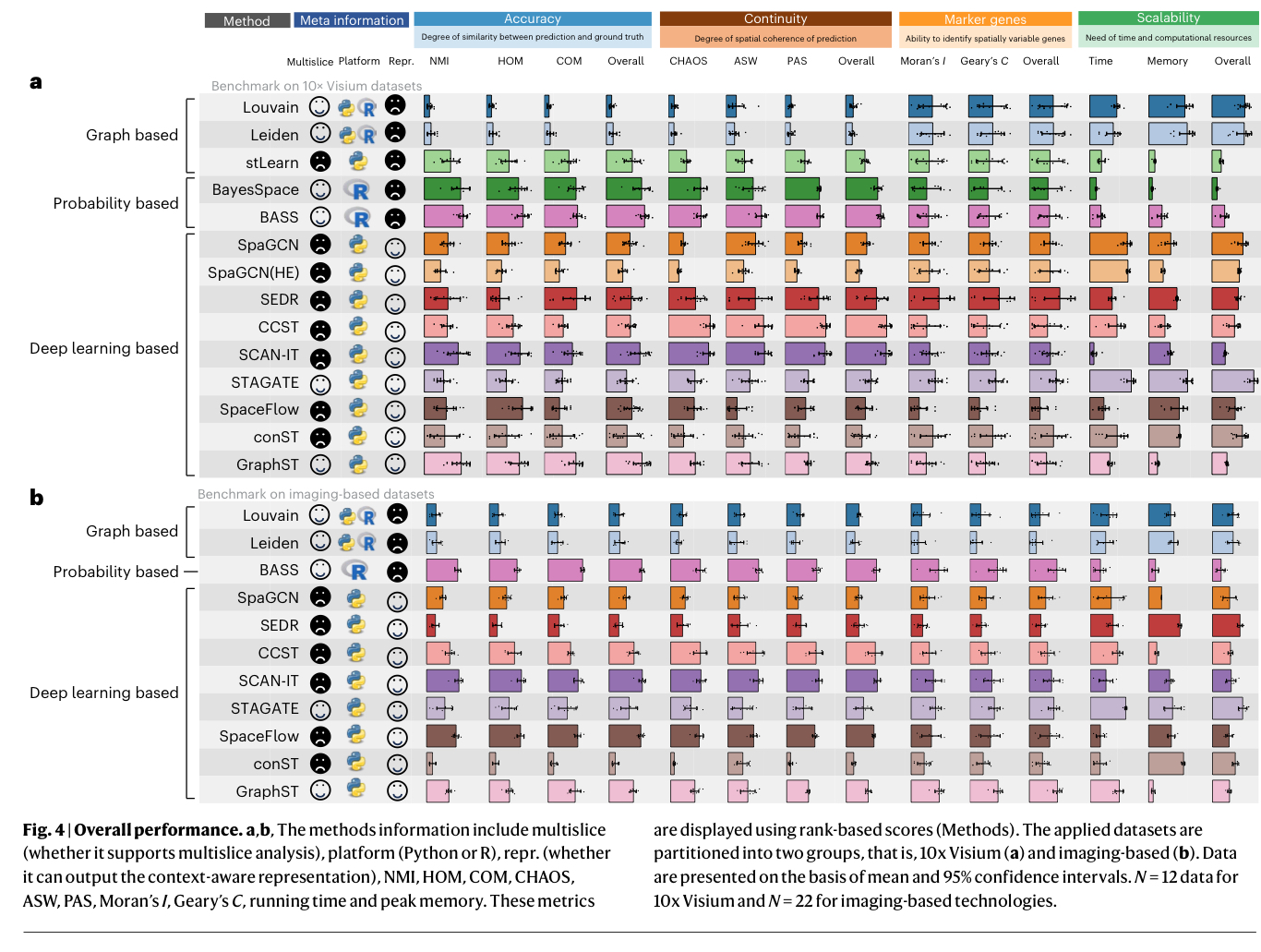
# Limitations
We found that all methods encounter challenges when faced with smaller, noncontinuous tissue domains
All methods encountered time and/or memory issues when applied to the dataset
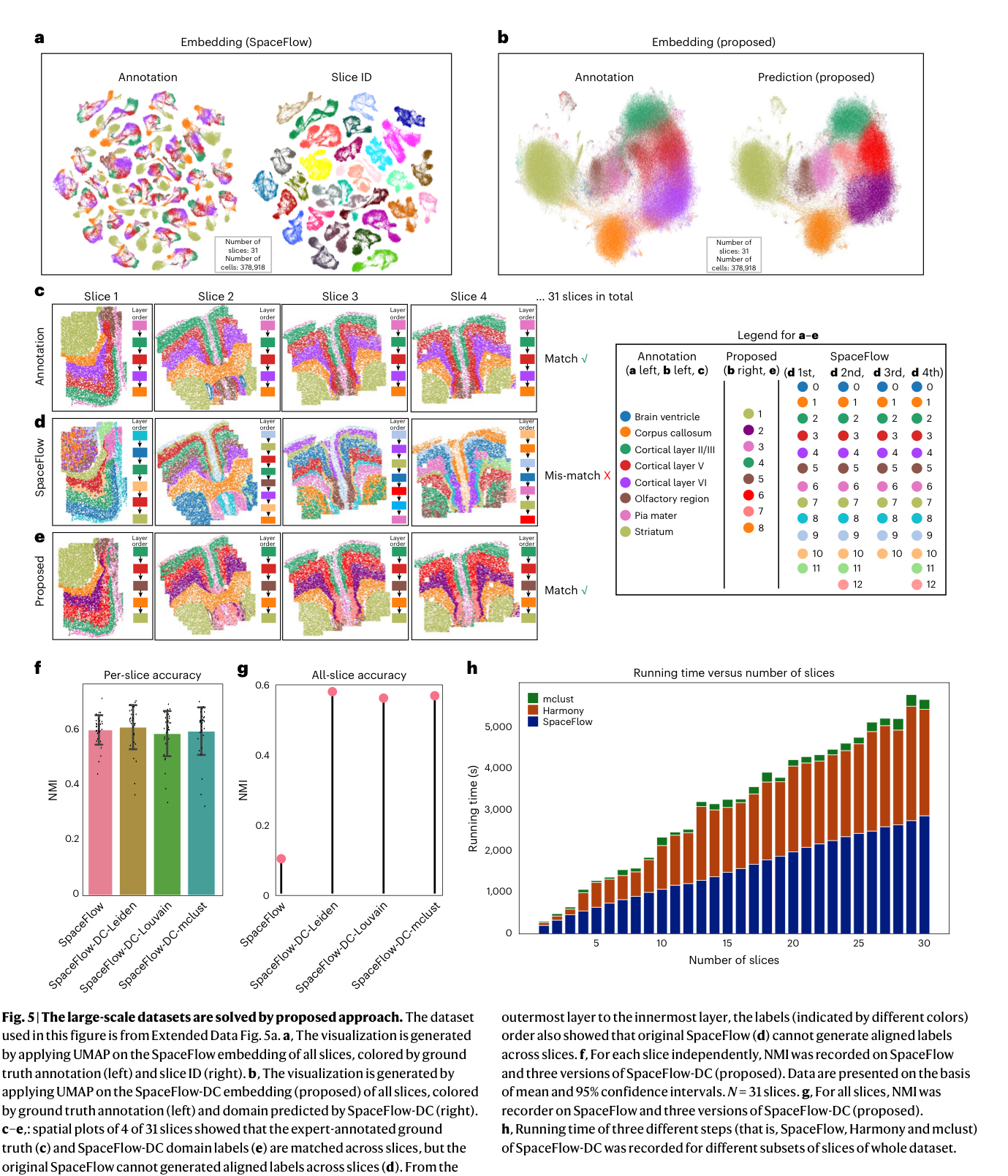
# A divide and conquer strategy enables large-scale scalability
使用的 base solver: SpaceFlow
这段话描述了一种 分而治之 的方法,应用于大规模的空间聚类问题。该方法基于 SpaceFlow 模型,结合了多种聚类算法和嵌入技术,以解决不同切片的空间数据分析,并整合它们的结果。
# 核心概念解释:
Divide and conquer strategy:分而治之策略将大规模问题拆解成多个小问题分别处理。在这里,原始数据是 31 个切片的空间数据,这些切片被分别处理,然后将结果进行整合。
SpaceFlow:这是一个用于单切片分析的工具,能够生成上下文感知的嵌入和空间聚类标签。每个切片独立运行 SpaceFlow,生成相应的嵌入和聚类结果。
UMAP 和批次效应:为了观察是否存在批次效应(不同批次之间的系统差异),他们将所有 31 个切片的嵌入拼接在一起,使用 UMAP 进行可视化,结果显示了批次效应的存在(见 Fig. 5a)。
Harmony 整合方法:为了解决批次效应,使用了 Harmony 这种整合方法。具体步骤是:首先用 PCA 将拼接后的嵌入降维至 30 维,然后应用 Harmony 对嵌入进行整合。整合后的嵌入被用来构建邻域图,随后用 UMAP 进行可视化,并用不同的聚类算法进行空间聚类。
空间聚类方法:使用了三种不同的聚类算法来生成空间聚类标签,包括:
- Leiden:一种社区检测算法。
- Louvain:另一种社区检测算法,类似 Leiden。
- mclust:基于高斯混合模型的聚类方法。
不同的组合方式被用于命名不同的方法,比如:
- SpaceFlow-DC-mclust:使用整合后的嵌入,结合 mclust 聚类方法。
- SpaceFlow-DC-Louvain:使用整合后的嵌入,结合 Louvain 聚类方法。
不同的算法对上下文感知表示的影响不大,可以根据需要选择不同的聚类算法且不会对最终性能产生显著影响
嵌入的可视化:有两种不同的嵌入可视化方式:
- Embedding (SpaceFlow):表示未经过 Harmony 整合的嵌入。
- Embedding (Proposed):表示经过 Harmony 整合的嵌入。
NMI(Normalized Mutual Information):为了评估聚类的准确性,使用了 NMI 指标。分别记录了每个切片的 NMI 和所有切片的整体 NMI。由于未对跨切片的 SpaceFlow 聚类标签进行对齐,导致每个切片的聚类准确性高,而整体的准确性较低。
# pre- and postpocessing
这段话讨论了在空间聚类分析中,预处理和后处理步骤对结果的影响,以及不同方法在不同数据特征下的鲁棒性(稳健性)。
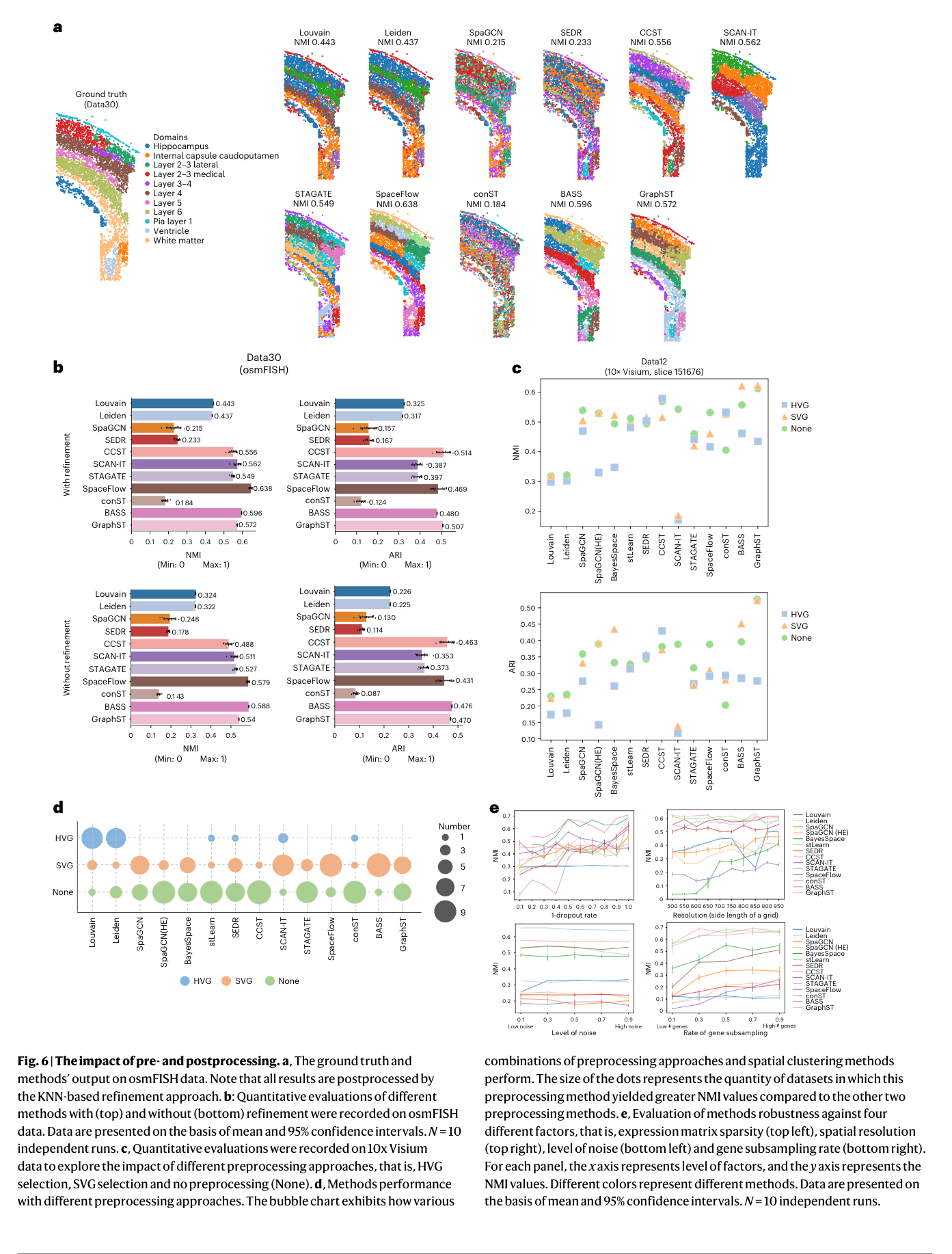
# 1. 预处理和后处理对性能的影响:
预处理步骤:在空间转录组数据的聚类分析中,预处理通常包括对基因进行选择,比如选择高度可变基因(HVG)或空间上可变基因(SVG),或者不进行任何选择。不同的预处理方式可能会影响聚类结果的质量。在实验中,他们测试了三种预处理方式:
- HVG 选择:选择高度可变基因。
- SVG 选择:选择空间上有差异的基因。
- 不选择基因:不进行任何基因选择。
通过分析,研究发现大约一半的方法在没有基因选择的情况下表现更好,表明预处理可能并非在所有情况下都有利。
后处理步骤:后处理主要用于对聚类结果进行精细化,比如通过 K 近邻(KNN)方法对空间域标签进行平滑处理,使得聚类结果在空间上更加一致。在这些实验中,发现所有方法在进行这种空间平滑的后处理步骤后,聚类性能都有所提升。
# 2. 数据特征对方法鲁棒性的影响:
空间转录组数据在不同实验中可能表现出不同的特性,比如基因表达矩阵的稀疏性、空间分辨率的高低、以及基因数量的多少。因此,模拟具有不同参数的数据有助于更广泛的参考,并且测试方法对这些特征的鲁棒性显得至关重要。
他们通过模拟数据,测试了不同方法对以下特征的鲁棒性:
基因表达矩阵稀疏性:即矩阵中缺失或零值的比例。在稀疏性增加时,大部分方法的性能下降,除了 CCST、Louvain 和 Leiden 这三种方法。
空间分辨率:即每个空间单位所包含的细胞或基因数目。大部分方法的性能在空间分辨率降低时有所提升,除了 Louvain、Leiden、SCAN-IT 和 SpaceFlow,这几种方法的性能在分辨率变化时表现稳定或下降。
基因数量:随着基因数量的增加,大多数方法的表现会提高,表明更丰富的基因信息有助于更准确的聚类。
噪声的影响:通过加入噪声测试方法的稳健性,结果显示,大多数空间聚类方法对噪声具有一致的鲁棒性,表明它们在噪声环境下的表现相对稳定。
# Discussion
这段话总结了在使用空间转录组数据进行空间聚类分析中对 13 种方法的基准分析,以及对未来基准研究设计的启示。
# 1. 研究概述:
- 目标:评估 13 种空间聚类方法,用于识别空间转录组数据中的空间域。
- 评价维度:包括准确性、连续性、标记基因得分(marker score)和可扩展性,为研究人员选择最优的聚类工具提供框架。
- 主要结论:没有单一方法在所有数据集上都表现最优,不同数据特征适合不同方法。此外,当前方法在识别小区域、多片段分析和处理大规模数据上存在局限性。
# 2. 方法的局限性与改进:
- 小区域识别困难:当前方法难以有效识别小的空间区域。
- 多片段分析能力欠缺:现有方法多局限于单片段分析,无法处理多个切片的数据。
- 可扩展性问题:随着空间转录组数据的规模增长,内存和计算时间的要求增加,现有方法在处理大规模数据时存在可扩展性不足的问题。
改进策略:提出了 “分而治之” 的策略,通过将数据分割并分别处理,再通过合并步骤提升大规模数据的处理能力。这种策略可以激励开发者结合现有工具,解决大规模空间数据集带来的挑战。
# 3. 额外数据的引入:
- HE 染色图像的影响:在评估 SpaGCN 方法时,研究发现加入 HE 染色图像并不总能提升性能,有时可能引入噪声。可能的解释是 HE 图像中包含的信息无法直接帮助识别组织结构,或是 HE 图像的建模方式仍有改进空间。
# 4. 研究的局限性与未来方向:
- 快速发展的技术领域:空间转录组学领域发展迅速,现有的研究可能未能涵盖最新的数据类型。
- 其他空间数据类型的缺失:研究主要集中在空间转录组数据,忽略了空间蛋白质组学和空间多组学数据,它们可能提供互补的信息。未来的研究应涵盖更多种类的数据和计算方法。
- 基准测试设计的启发:研究强调了未来基准测试应考虑的数据集多样性、可靠的 “ground truth” 注释,以及更广泛的空间数据类型,以确保研究结果的普适性和可比性。
# 5. 评价指标:
- 相似度指标:衡量方法预测结果与 “ground truth” 之间的相似度。例如,研究中使用 NMI、HOM 和 COM 来评估这种相似性。
- 空间连续性:如 CHAOS、PAS 和 ASW 指标,用于衡量预测的空间域的连续性。然而,这类指标并非总能代表好的预测结果,因为高空间连续性不一定意味着高准确性。
- 可扩展性:运行时间和内存占用是衡量方法可扩展性的重要指标,尤其是在大规模数据集上。
- 数据集选择:优先选择带有可靠 “ground truth” 的数据集。如果没有可靠的真实数据集,模拟数据可作为替代品。
# Method
we adopted two approaches based on the capabili- ties of the methods.
- For algorithms where we could directly input the expected number of spatial domains (for example, SpaGCN and BayesSpace), we set the parameter to the actual number of domains obtained from our ground truth.
- For those algorithms that could only accept clustering resolution, we searched the resolution that best matches the expected number of spatial domains.
# 空间聚类方法:
Louvain:一种非空间聚类算法,基于模块度优化,通过反复合并和分割社区来分配每个点,直到得到期望的聚类结果。
Leiden:类似于 Louvain 的非空间聚类算法,但进一步改进,可以通过合并相似社区来增强聚类效果。
SpaGCN:利用图卷积网络(GCN)将基因表达、空间位置和组织学信息整合起来,通过无监督的迭代聚类划分空间域。用于无图像数据的情况。
SpaGCN(HE):类似于 SpaGCN,但适用于有 H&E 组织学图像的数据,通过将 RGB 值转换为 3D 坐标以计算空间点之间的欧几里得距离。
BayesSpace:贝叶斯统计方法,利用基因表达矩阵的低维表示进行空间聚类,通过空间先验鼓励相邻点属于同一簇。适用于 “spot” 形式的空间数据。
stLearn:基于空间形态学基因表达(SME)归一化数据进行无监督聚类,将相似点归为同一簇,并利用空间信息识别组织内的子类群。需要组织学图像作为输入。
SEDR:使用深度自编码器构建基因表达数据的低维表示,然后与空间信息结合,通过变分图自编码器进行同时的空间嵌入和聚类。
CCST:将细胞位置和基因表达信息编码为邻接矩阵和基因表达矩阵,通过深度图信息最大化网络获取包含空间和基因表达信息的节点嵌入,并通过主成分分析(PCA)和 k-means++ 进行聚类。
SCAN-IT:使用图像分割方法处理空间聚类问题,将细胞视为图像中的像素,基因表达视为类似 RGB 通道的数据,构建几何感知的空间邻接图,利用深度图信息最大化生成低维嵌入进行聚类。
STAGATE:将空间位置信息转化为空间邻居网络,并结合基因表达信息,通过图注意力自编码网络生成低维嵌入,用于空间聚类。
SpaceFlow:利用深度图神经网络(GNN)将基因表达相似性和空间信息融合,通过空间正则化生成空间一致的低维嵌入,用于聚类。
conST:基于对比学习方法,整合多模态的空间转录组学(SRT)数据(如基因表达、空间信息和形态学),通过数据增强和三层对比学习生成低维嵌入进行聚类。
BASS:一种多尺度、多样本的空间转录组学数据分析方法,采用贝叶斯分层建模框架进行聚类。
GraphST:通过图自监督对比学习模型进行空间聚类,结合 GNN 和自监督学习,学习空间转录组数据中的基因表达和空间位置信息嵌入,用于聚类。
这些方法都致力于结合空间位置信息和基因表达数据,通过不同的模型和算法来提高空间聚类效果。
# Simulated data
分别考察了不同的 spatial resolutions\expression matrix sparsity\gene subsampling rate\various levels of noise, 使用了不同的方法
# Benchmark metrics
# NMI
衡量两个聚类之间的相似性,NMI 属于 0-1 之间,越接近 1 说明越相似。
衡量的是从一个聚类结果预测另一个聚类结果所需的额外信息量
适用于类别数量不一致的聚类结果评估
\begin{align*} NMI = \frac{MI(P,T)}{\sqrt{H(P)H(T)}} \end{align*}Suppose P is the spatial domain clustering result, T is the ground truth clustering label, their entropies are H(P) and H(T) , respectively, and the mutual information is MI(P,T), then NMI is computed as:
# ARI
比较两个聚类结果种每一对数据点的划分是否一致
适合在类别数量一致的情况下评估两个聚类结果
ARI is computed as:
# HOM(Homogeneity Score):
衡量聚类结果与已知真实标签相比的同质性。如果一个聚类的每个簇只包含属于同一类别的数据点,则聚类是同质的。HOM 得分范围为 0 到 1,1 表示完全同质。
# COM(Completeness Score):
评估聚类结果相对于真实标签的完整性。如果属于同一类别的所有数据点都被正确分配到同一簇,则聚类是完整的。COM 得分范围为 0 到 1,1 表示完全完整。
# CHAOS
CHAOS(Continuity Assessment for High-throughput Open Science):用于评估质谱成像和空间转录组学领域中空间连续性的指标。CHAOS 得分越低,表示空间域识别的连续性结果越好。计算过程包括以下步骤:
- 构建每个数据的 1 - 最近邻(1-NN)图,每个细胞与在物理空间中具有最小欧几里得距离的其他细胞连接。
- 定义连接的权重 \( w_{kij} \):如果细胞 \( i \) 和细胞 \( j \) 在簇 \( k \) 中相连,则 \( w_{kij} = d_{ij} \)(它们之间的欧几里得距离),否则为 0。
- CHAOS 得分计算公式为:\begin{align*} CHAOS = \sum_{k=1}^{K} \sum_{i=1}^{n_k} w_{kij} \end{align*} 其中 \( n_k \) 是簇 \( k \) 中的细胞数量,\( n \) 是数据中的总细胞数,\( K \) 是唯一空间域的数量。
# PAS(Percentage of Area Shared):
用于量化空间转录组学中空间域识别算法的空间同质性指标。较低的 PAS 分数表示检测到的空间域的连续性更好,期望在空间域内细胞的同质性更高。PAS 分数的计算方式是:具有不同空间域标签的细胞占其邻近十个细胞中至少六个的百分比。
# ASW(Average Silhouette Width):
最初用于评估聚类标签与嵌入(或距离矩阵)之间一致性的指标。ASW 被扩展用于评估预测空间域相对于物理空间的空间一致性。ASW 的值范围为 - 1 到 1(通常重标定到 0-1),ASW 值越接近 1,性能越好。ASW 的计算需要先定义轮廓宽度(Silhouette Width,SW),SW 的计算方式如下:
SW 的定义:
- 设 \( a \) 为细胞与同一空间域内所有其他细胞的平均距离。
- 设 \( b \) 为细胞与下一个最近聚类中所有其他细胞的平均距离。
- 则细胞的 SW 计算公式为:
ASW 的计算:ASW 通过对所有细胞的 SW 值取平均来获得。
# Moran's I
是一种用于量化空间自相关程度的指标,广泛应用于空间统计学,尤其是在空间组学领域评估检测到的 SVG(空间变异基因)是否表现出有序的空间表达模式。Moran's I 的值范围为 - 1 到 1:
- 接近 1:表示存在明显的空间基因表达模式,说明相似的基因表达值在空间上是聚集的。
- 接近 0:表示随机的空间基因表达模式,说明基因表达在空间上没有显著的规律性。
- 接近 - 1:表示基因表达模式呈现出棋盘状的分布,即相似和不同的表达值交替分布。
在计算 Moran's I 时,假设有两个细胞 \(x_i\) 和 \(x_j\) 的基因表达值,\(\bar{x}\) 是该基因的平均表达值,\(N\) 是细胞的总数。Moran's I 的计算公式为:
\begin{align*} & Moran's \ I = \frac{N}{W} \cdot \frac{\sum_{i=1}^{N} \sum_{j=1}^{N} w_{ij} (x_i - \bar{x})(x_j - \bar{x})}{\sum_{i=1}^{N} (x_i - \bar{x})^2} \\ & where \\ & w_{ij} = \begin{cases} 1,& \text{ifi and j are spatial neighbors} \\ 0,& \text{else} \end{cases} \\ & where \\ & W = \sum_{i,j}{} w_{ij} \end{align*}其中:
- \(W\) 是所有权重 \(w_{ij}\) 的总和,通常表示细胞之间的空间权重。
- \(w_{ij}\) 是细胞 \(i\) 和细胞 \(j\) 之间的空间权重,反映它们在空间上的关系(例如,距离或邻接关系)。
# Geary's C
是一种用于量化空间自相关程度的指标,类似于 Moran's I,但它的计算方式和数值范围有所不同。以下是 Geary's C 的主要特点和计算方法:
- 比较:Geary's C 和 Moran's I 都用于分析空间自相关,但 Geary's C 更加关注相邻细胞之间的差异性,而 Moran's I 更加关注整体的聚集性。
计算方法
在计算 Geary's C 时,假设使用与 Moran's I 相同的符号表示,计算公式为:
\begin{align*} & Geary's \ C = \frac{N \cdot \sum_{i=1}^{N} \sum_{j=1}^{N} w_{ij} (x_i - x_j)^2}{2W \cdot \sum_{i=1}^{N} (x_i - \bar{x})^2} \end{align*}其中:
- \(N\) 是细胞的总数。
- \(w_{ij}\) 是细胞 \(i\) 和细胞 \(j\) 之间的空间权重(通常基于距离或邻接关系)。
- \(x_i\) 和 \(x_j\) 是细胞 \(i\) 和细胞 \(j\) 的基因表达值。
- \(\bar{x}\) 是该基因的平均表达值。
# 代码实现
# 主代码
1 | import scanpy as sc |
# 调用的代码
# prefilter_genes
1 | def prefilter_genes(adata,min_counts=None,max_counts=None,min_cells=10,max_cells=None): |
对于 sc.pp.filter_genes 的解释:
数据预处理
这里介绍一下 scanpy 中常用的组件
pp: 数据预处理
tl: 添加额外信息
pl:可视化
1 | sc.pl.highest_expr_genes(adata, n_top=20) # 每一个基因在所有细胞中的平均表达量(这里计算了百分比含量) |
# prefilter_specialgenes
1 | def prefilter_specialgenes(adata,Gene1Pattern="ERCC",Gene2Pattern="MT-"): |
过滤掉以 ERCC 和 MT - 开头的基因
- "ERCC" 是指外源对照基因(External RNA Controls Consortium),通常用于实验控制。
- "MT-" 是指线粒体基因(Mitochondrial genes),通常用于质量控制。
# search_l
1 | def search_l(p, adj, start=0.01, end=1000, tol=0.01, max_run=100): |
# search_res
1 | def search_res(adata, adj, l, target_num, start=0.4, step=0.1, tol=5e-3, lr=0.05, max_epochs=10, r_seed=100, t_seed=100, n_seed=100, max_run=10): |
# SpaGCN 里面的 train 函数
1 | def train(self,adata,adj, |
# predict
1 | def predict(self): |